

Introduction
Materials and Methods
Description and collection of diseased samples
Library Sequencing
Bioinformatics and statistical analysis
Results
Bacterial diversity
Characteristics of the bacterial community
Comparing bacterial genera in the SCD and MCD groups
Discussion
Introduction
The citrus fruit is a classic example of an orchard fruit, as it is low in carbs, moderate in protein and fat content, and very high in micronutrients. Citrus crops are not considered to be a staple fruit source (Kreitzman et al., 2020). According to the China Statistical Yearbook 2021, Guangxi's citrus output ranked first in China, reaching 13.821 million tonnes. Additionally, the province's distinctive taste is well loved and recognized throughout the nation (http://www.stats.gov.cn/, accessed November 25, 2022). Citrus is an economically important crop, but its production is fraught with difficulties, including inadequate soil nutrients, improper irrigation, and a wide range of soil- and plant-borne illnesses. Since the 1930s, Zhejiang, Hunan, Fujian, Guangxi, and other provinces have seen outbreaks of citrus canker disease caused by Xanthomonas citri pv. Citri (henceforth referred to as Xcc), a significant quarantinable disease harming citrus output (Liao and Huang, 2021).
Microorganisms found in plant tissues and organs are known as endophytes, and the corresponding microenvironments may be further subdivided. The community structure and roles of endophytic bacteria and their host plants evolve as a result of mutual adaptation and interaction in response to stress and environmental stressors (Azarbad et al., 2016; Santos-Medellín et al., 2017; Zhao et al., 2022). Several studies have demonstrated the effects of various factors that influence endophytic bacterial communities in host tissues. For example, differences in management methods can lead to changes in the fungal endophytic communities of grass species (Wemheuer et al., 2019). The proximity of trees to sewage outfalls increases the diversity of bacterial endophytes in their roots (Janowsky et al., 2019), and the core microbiota differ between juvenile and mature leaves of Pinus flexilis (Carper et al., 2018). The abundance of fungal communities in Populus angustifolia twigs varies by genotype (Lamit et al., 2014). Additionally, endophytic bacterial communities change with biotic constraints on plants.
Studies have shown that insect galls affect the formation of fungal endophyte communities in plant leaves (Fernandez-Conradi et al., 2019). The plant microbiome is dynamic, and pathogens invading plants can cause dysbiosis in bacterial community, leading to reduced plant resilience and ultimately disease (Chen et al., 2020). At the same time, plants enlist the help of the microorganisms that are in their immediate environment in order to create a new equilibrium that is better able to combat or withstand the effects of the stress caused by pathogens (Liu and Brettell, 2019; Rizaludin et al., 2021). This strategy is used by microorganisms to suppress pathogens in the soil, thereby facilitating plant growth (Weller et al., 2002). It has also been shown that plants can recruit specific microorganisms through the release of chemicals to form microbial communities to adapt to adversity (Schulz-Bohm et al., 2018; Voges et al., 2019). One key element in this response is the production of secondary metabolites, such as flavonoids. Flavonoids are chemicals that play a multifaceted role in plant homeostasis and are essential in facing biotic and abiotic stresses to sustain plant growth. These compounds act as signaling molecules and antioxidants, helping the plant adapt and survive in challenging conditions. They can also influence interactions with microorganisms in the soil, contributing to the plant's overall stress response. The structure of the flora that is created by a plant after it has been infected by pathogenic bacteria may be connected to the degree to which the plant has been infected by the bacteria in a back-and-forth competition. Few reports exist regarding how citrus canker affects endophytic bacterial populations. To gain insight into the relationship between citrus canker disease and endogenous microbial diversity, a community of endogenous bacteria in citrus canker disease plants at different infection stages was investigated and analyzed using high-throughput sequencing libraries based on 16S rRNA.
Materials and Methods
Description and collection of diseased samples
Although orchards in Bayan, Nanning, Guangxi Zhuang Autonomous Region, China, are managed in a unified and scientific way, they can still become infected by citrus canker disease. We randomly selected an area and collected leaves with citrus canker disease from different citrus trees, grouping the samples based on disease stage differences. The samples are all from the same variety of citrus (Orah), and the moderate citrus canker disease (MCD) group represented the early stage of citrus canker infection, with the leaves showing yellow or dark yellow-green oil-like small spots and beige round disease spots. The severe citrus canker disease (SCD) group represents the late stage of citrus canker infection, in which the disease spot epidermis is ruptured and woody embolized, light brown, and gray-white in the center (Fig. 1). Two groups of leaves were transported in sterile bags from the sampling site to a laboratory along a cold chain, with three separate samples from each group.

Library Sequencing
The samples were transported to the laboratory through the cold chain, and after disinfecting the surfaces of the leaves, DNA was extracted from healthy parts near the damaged leaves using a DNA extraction kit (MN NucleoSpin 96 SOIL). The V3-V4 regions of 16S rRNA amplification primers were 335F (CADACTCCTACGGGAGGC) and 769R (ATCCTGTTTGMTMCCCVCRC). The amplified DNA was purified and sequenced at BioMarker Technologies, Inc. Then, the data were stored at CNGB (CNP0003590, https://db.cngb.org/).
Bioinformatics and statistical analysis
The raw data were spliced using FLASH v1.2.11 and then quality filtered, and chimeras were removed (Trimmomatic, version 0.33; UCHIME version 8.1) (Edgar et al., 2011; Magoč and Salzberg, 2011; Bolger et al., 2014). The sequences were 97% matched (USEARCH version 10.0) and externally filtered at a threshold of 0.005%for all sequences (Bokulich et al., 2012; Edgar, 2013). The SILVA database was selected for species annotation of the resulting sequences (Quast et al., 2013).
A microbial diversity analysis was conducted through a bioinformatics analysis using the BMKCloud tool (http://www.biocloud.net/). This tool can help us directly use sequencing databases to compare and analyze species diversity (Luo et al., 2020; Zhang et al., 2022). The Alpha Diversity Index of each sample (Simpson, Shannon, and Coverage) was evaluated using the “picante” package in R (v3.1.1), after which significant differences between the groups were tested with Student’s t-tests. QIIME2 software was used to perform a beta diversity analysis, and the distances between the samples were calculated using the weighted unifrac algorithm to obtain PCoA analysis results. APERMANOVA analysis could test whether the beta diversity between the samples of different groups differed significantly.
Results
Bacterial diversity
A total of 480,637 pairs of reads were acquired from the sequencing of six samples based on the 16S rRNA sequencing analysis. In total, 478,651 clean reads were produced after a sequencing data quality assessment, with an average of 79,610 clean reads produced for each sample. There was between 51% and 55% GC. MEGAN software was then used to categorize the samples further following the BLAST findings (Huson et al., 2007). The coverage of all samples exceeded 99%, suggesting that the majority of bacteria in the sample are represented in the detected sequencing findings (Suppl. Table 1). There are more types of bacteria to look for in the MCD group, as indicated by the higher Shannon index and higher Simpson index of diversity 1-D (Fig. 2). The Student’s t-test indicates significant differences between the two groups (Table 1).
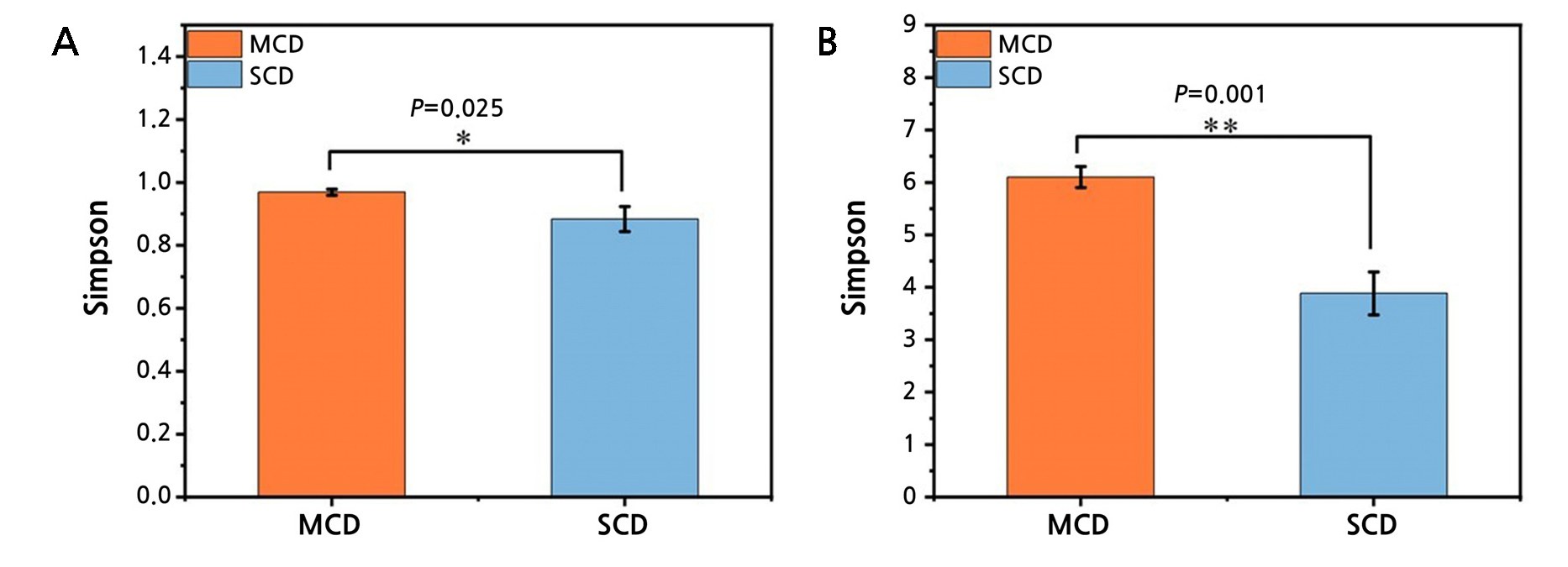
Table 1.
Student's t test results for significance of the alpha diversity index
| Group (Mean ± SD) | t | p | ||
| MCD (n = 3) | SCD (n = 3) | |||
| Simpson | 0.97 ± 0.01 | 0.88 ± 0.04 | 3.480 | 0.025 |
| Shannon | 6.11 ± 0.20 | 3.88 ± 0.41 | 8.466 | 0.001 |
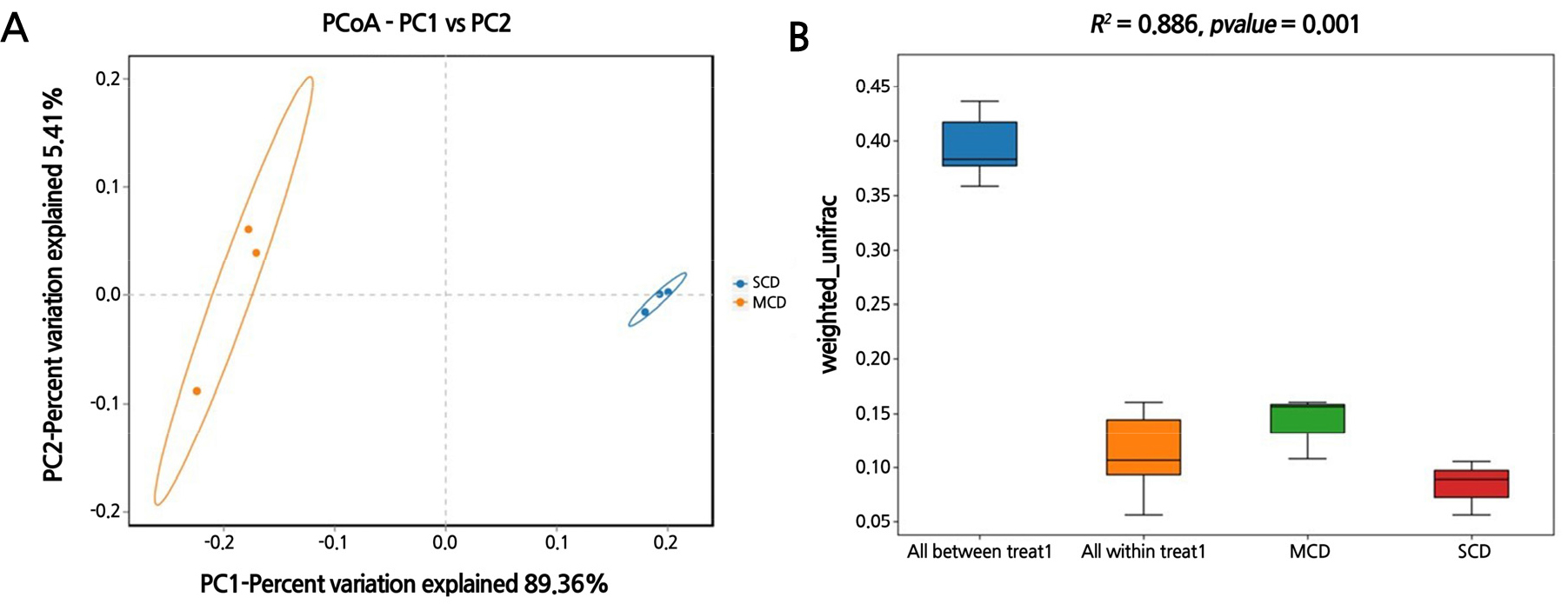
Bacterial communities in the SCD group were found to be geographically closer to one another and more similar to one another than those in the MCD group (Fig. 3A). The results of the PERMANOVA analysis indicated significant differences in the microbial communities of leaves infected with citrus canker at different stages (p = 0.001), contributing 88.6% (R2 = 0.886) to the total variation of the community (Fig. 3B).
Characteristics of the bacterial community
Based on the taxonomic categorization, every single 16S rRNA gene segment that was found in either set of samples was determined to belong to bacteria. These pieces were then categorized further into recognized bacterial phyla for the MCD and SCD groups to which they belonged.
It was discovered that Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes were the main bacterial groupings in both the MCD and SCD groups (Fig. 4). In both groups, Proteobacteria were the most prevalent phylum, accounting for 75.8% and 62.4% of the total 16S rRNA gene fragments, respectively. In the MCD group, Firmicutes were found in very high numbers (relative abundance of 25.2%), but in the SCD group, the phyla were only found in very small numbers (0.55%). Actinobacteria, on the other hand, only made up 2.4% of the MCD group but 15.6% of the SCD group. Bacteroidetes were responsible for 7.2% of the MCD group and 8.0% of the SCD group. Less than 1% of the remaining phyla were identified.
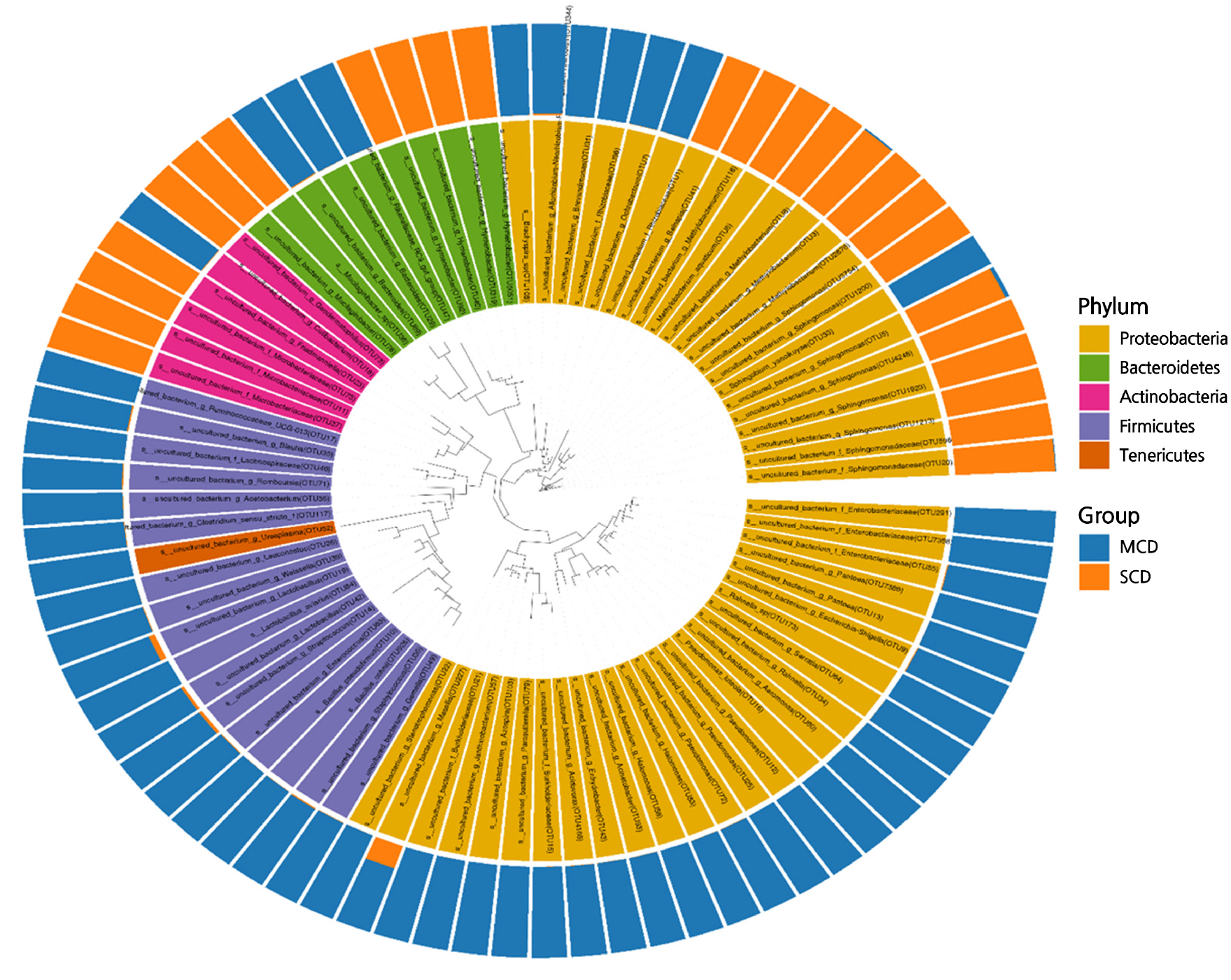
The number of reads identified by sequencing varies from level to level, and as the number of classification levels increases, some reads from the upper level may not be accurately assigned to the lower level. This is an essential point to keep in mind. Alphaproteobacteria was the class of Proteobacteria that was found to be most abundant in the SCD group (74.8%), also accounting for 18.2% of the total Proteobacteria in the MCD group). This was followed by Gammaproteobacteria (0.7 and 43.6%, respectively) and Deltaproteobacteria (0.3 and 0.6%, respectively). Both the Rhizobiales (38.2 and 13.9% of the total order, respectively) and Sphingomonadales (35.5 and 1.7% of the entire order, respectively) orders occurred in both the SCD and MCD groups at the order level. These two orders are part of the Alphaproteobacteria class of bacteria.
Comparing bacterial genera in the SCD and MCD groups
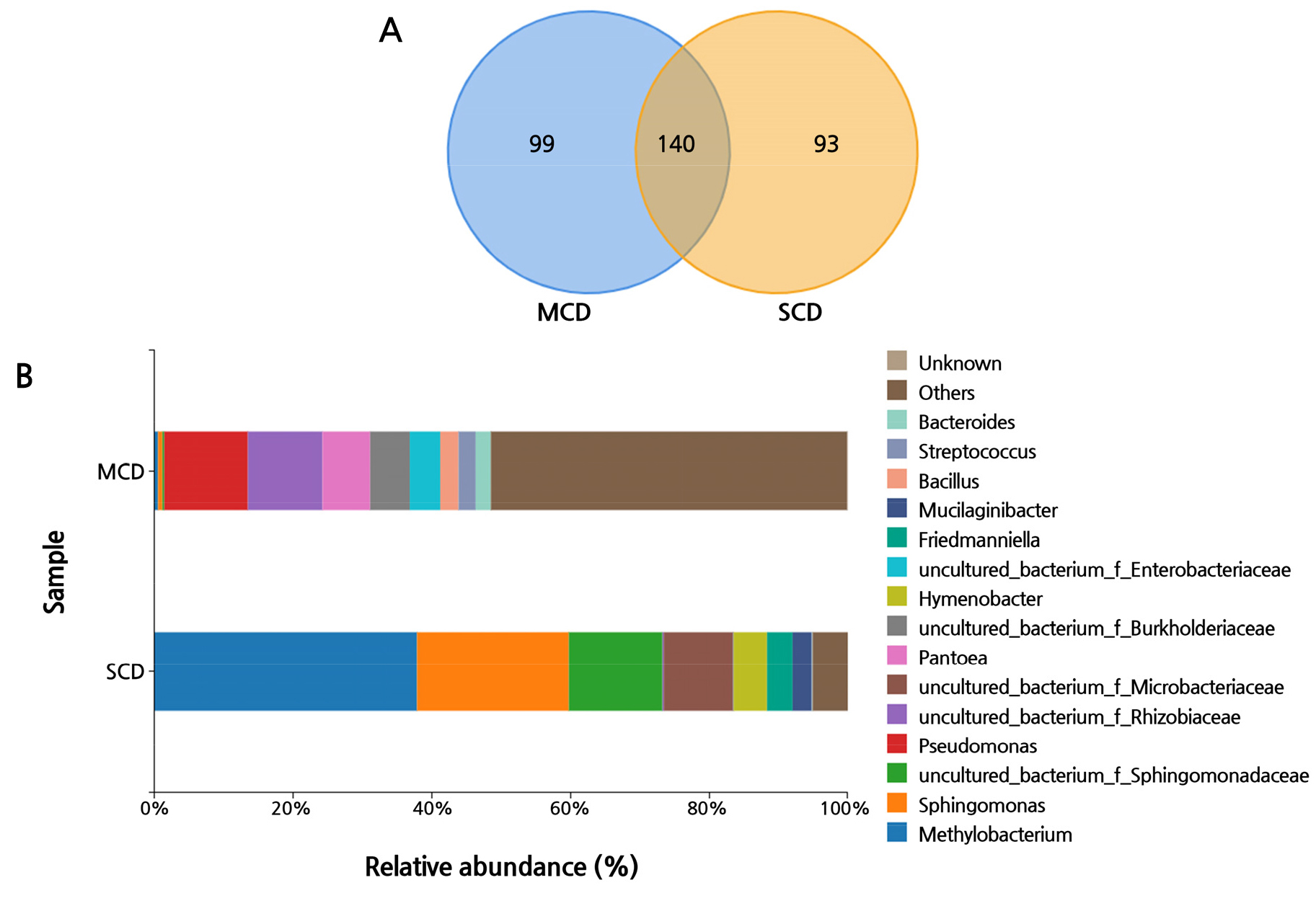
In the next step of the process, a Venn diagram was devised by taking into account bacterial populations at the genus level and using a confidence threshold cut-off of more than 97%. The number of systematic identifications at the genus level was comparable across the two groups; however, the percentage distributions of each genus within the respective group differed substantially. In accordance with the Venn diagram, the two groups contain the same 140 genera (Fig. 5A). In the SCD group, there were nine genera with more than 1% each, which accounted for 96.8% of the total, and in the MCD group, there were 21 genera, which accounted for 64.6% of the total, consistent with the findings of the alpha diversity measure.
The relative abundance of the top 15 genera in both the SCD and MCD groups is shown in Fig. 5B. There are a significant number of operational taxonomic units (OTUs) that are not identified at the genus level in this image. This has led the researchers to the conclusion that the microbiome in such samples, which has not been formally annotated, has a great multitude of potentially unique microbial resources. In order to uncover these resources, researchers will need to put in additional efforts. In the SCD group, most of the reads were annotated to the genus Methylobacterium (37.8% of the total genera), followed by the genera Sphingomonas (21.9%), uncultured_bacterium_f_Sphingomonadaceae (13.5%), and uncultured_bacterium_f_ Microbacteriaceae (10%). On the other hand, the MCD group only had a detection rate of 0.6% for Sphingomonas and 0.5% for Methylobacterium. Other genera are more equally distributed, including uncultured_bacterium_f_Rhizobiaceae (10.7%), Pantoea (6.9%), and Bacillus (2.6%). Pseudomonas accounted for the greatest proportion of the overall genera (about 12%).
When compared to the MCD group, the SCD group had significantly more Sphingomonas and Methylobacterium in their systems. The data given in this article provide undeniable evidence to back this conclusion. However, the data show that the bacterial communities of the SCD and MCD groups were regarded to be separate from one another and were thought to be unique from one another as well. According to the findings of a study conducted by Trivedi, the Huanglong illness causes changes in the microbial community that are connected to citrus roots, leading to an increase in the prevalence of both Methylobacterium and Sphingobacterium (Trivedi et al., 2010). In addition, the MCD group was shown to have a much higher abundance of Pseudomonas and Bacillus than the SCD group. It has been observed that native Bacillus and Pseudomonas have the capacity to inhibit Xcc, which has been proven in certain studies to lead to a decrease in the bacterial variety present in plant leaves. According to the findings of certain studies, an Xcc infection may be one factor contributing to this decline (Murate et al., 2015; Michavila et al., 2017; Jelušić et al., 2021).
Discussion
The phytobiomeconsists of plants, their environment, and a variety of interacting microscopic and macroscopic animals. Together, these factors impact plant health and productivity (Leach et al., 2017; de Souza et al., 2020). Due to the limitations of culture conditions and methods, microorganisms identified by traditional methods cannot truly reflect the structure of the plant endophytic bacteria community. Through the use of Illumina high-throughput sequencing of the V3-V4 hypervariable region of bacterial 16S rRNA genes, the complex bacterial communities connected to the phyllosphere of citrus were thoroughly characterized and compared in this work.
According to the findings of our research, severe symptomatic phyllosphere samples had a direct pathogenic influence on reducing the overall bacterial richness, which is also referred to as alpha diversity. When compared to moderately symptomatic phyllosphere samples, this result was obtained. Other studies have confirmed this pattern; after the fruit and leaf tissues of bell pepper were infected with fungal pathogens, the species richness and diversity of fungi in the infected tissue samples were reduced, whereas uninfected stem tissues were not affected (Liu et al., 2022). Manching hypothesized that the action of Cochliobolus heterostrophus was responsible for the decrease in the number of bacterial species and the variety of those species in the maize leaf sphere (Manching et al., 2014). A study showed that bananas that were not infected with bacterial wilt disease had a richer microbial community (Suhaimi et al., 2017), and another study showed that on average, the overall microenvironment studied (rhizospheres, rhizospheres, pseudostems and leaves of banana), when infected by Fusarium oxysporum f. sp. Cubense (FOC), have lower diversity levels than healthy plants (Köberl et al., 2017). There are a number of hypothesized causes of this phenomenon, one of which is that the pathogenic bacteria and the afflicted plants engage in a struggle for nutrition supplies and survival locations, which leads to decreased microbial diversity in infected plants (Bulgari et al., 2011).
Alterations of the species diversity of microbial communities are among the aspects that can be utilized to diagnose biological dysregulation. There were noticeable and substantial changes between the groups’ endophytic bacterial communities. Chen's research proves that plants have evolved a genetic network to regulate the homeostasis of microbial flora to maintain plant health (Chen et al., 2020). At the same time, these changes are very similar to how the human genetic network is regulated (Sokol and Seksik, 2010; Turpin et al., 2018). According to the taxonomic categorization, the majority of the bacteria found in citrus leaves belong to the phyla Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria; these findings support the conclusions reached in earlier work (Yan et al., 2021). In different stages of a citrus canker disease infestation, the taxonomy of species annotation showed that the dominant species resources of the MCD group and the SCD group were inconsistent at the phylum level. It is speculated that plants may have attempted to maintain plant homeostasis in different ways in the early and/or late stages after becoming infected with citrus canker disease.
Through a variety of mechanisms, including the synthesis of antimicrobial peptides, bacteriolysis, and niche competition, some species of the phylum Firmicutes have the ability to suppress Xcc, the agent responsible for citrus canker disease (Islam et al., 2019; Rabbee et al., 2019; Rabbee et al., 2022; Wang et al., 2022). According to the findings of the classification, for the leaves of the SCD group that had been severely infected with Xcc, Firmicutes were in a position to compete with one another and interacted with pathogens to ward off the invasion of the pathogens, which led to a significant decrease in the population. As a result, differences in the composition and variation trends of endophytic bacteria that are attributable to Firmicutes would be a reflection of the prevalence of citrus canker disease. In other words, the dynamics of leaf endophytic bacterial populations may predict the pathways that citrus will take to combat canker disease.
Throughout their lives, plants are susceptible to stress from different sources, and when they are stressed, several defense mechanisms are activated to cope with the stress. For instance, in response to one type of pathogen (Rhizoctonia solani), the genes of essential defense hormones (SA, JA, and ET) of plants are increased in Sphagneticola trilobata (Qi et al., 2022). Plants build defense mechanisms to help them adapt to salt stress by coordinating the production, signaling, and metabolism of certain different hormones (Yu et al., 2020). Particularly, after plant stress, a rise in the symbiotic microbe population is seen, with the term “Defense Biome” suggested in relation to this (Liu et al., 2020).
Plant-related microorganisms can adjust the plant structure in time to bring benefits to the plants when they respond to biological or abiotic stressors (Wang and Song, 2022). Therefore, when plants are under assault by diseases, they can “cry for help” from the microbiota that are natural to their environment (Bakker et al., 2018). New research suggests that plants recruit large numbers of microorganisms when they are stressed and that their presence is likely to be beneficial to their survival. For instance, when sugar beetroots are subjected to the fungal disease Rhizoctonia solani, they attract members of the bacterial families Burkholderiaceae, Sphingobacteriaceae, and Sphingomonadaceae into the endosphere. This has the effect of suppressing the fungal infection (Carrion et al., 2019). Another study showed that the selective recruitment of new bacteria from Arabidopsis roots after leaf infestation with Hyaloperonospora arabidopsidis provides disease-resistant and growth-promoting benefits to plants (Berendsen et al., 2018).
The endophytic community tended to favor growth and stress-resistance microorganisms as leaf infection levels rose, while the fraction of bacteria that actively combated pathogens fell. The evolutionary trend and/or direction of the endophytic community tended towards “weakening its spear and strengthening its shield.” It should also be noted that in the SCD group, Actinobacteria and Proteobacteria accounted for more than 90% of the total, and the homogeneity of the community was dramatically decreased in comparison with the MCD group. Bioactive compounds such as alkaloids, phenols, flavonoids, and peptides are secreted by most endophytic Actinobacteria; these chemicals have antibacterial activities and have widespread medical and therapeutic use (Xu et al., 2014; Weber et al., 2015; Zin et al., 2017). Sphingomonas and Methylobacterium belong to the phylum Proteobacteria, these two are beneficial for plant survival (Asaf et al., 2020; Zhang et al., 2021).
Transplanting endophytic bacteria to leaves growth-promoting plant hormones are released as a consequence of Sphingomonas' regulation of osmotic pressure, which protects plants (Khan et al., 2014; Asaf et al., 2017). Sphingomonas can be used as a biofertilizer to improve the yield and qualityof plants (Asaf et al., 2018) and may also play an important role in environmental remediation in the future (Asaf et al., 2020). Methylobacterium can enhancethe nutrient transportation in leaves and it can be use the methanol that released by plants to produce products that promote healthy plant growth (Bishop et al., 2011; Fatin Nabilah et al., 2021), which is an important subject of bioengineering research (Zhang et al., 2021). With only two bacteria making nearly 60% of the genus in the SCD group, only a few members of the microbiota in the endophytic leaf layer may be involved in the response to an Xcc infection. Thus, the “cry for help” method is utilized by the surrounding symbiotic microorganisms when the citrus leaf is breached by an adversary (citrus canker disease), restoring the leaf to its natural condition.
A comparison was conducted of endophytic bacterial communities of citrus leaves at different stages of an Xcc infection, and the results showed that compared to the SCD group, the MCD group had more abundant and diverse bacteria. We found that as citrus canker disease continues to develop and the infection intensifies, the composition of the endophytic microbial population in the leaves will also change. In addition, when the leaves are infected with citrus canker disease up to the later stages, the number of Firmicutes decreases significantly. However, the increases in Sphingomonas and Methylobacterium compared to the MCD group have a positive effect on most plants.
This article provides new evidence of the “cry for help” strategy and provides a new breakthrough point for finding plant biological control solutions. That is to say, not only can we screen for biocontrol strains with stronger antagonistic abilities from plant tissues infected with citrus canker disease at different stages, but in the future, sequencing technology can also be used to focus on studying the functions of microbial communities, combined with other omics to elucidate the mechanisms of plant and pathogen occurrence. Another method of creating plant biological control technologies involves the application of advantageous strains or artificially formed microbial communities to alter the microbial structure and function in order to prevent or minimize infections by harmful bacteria.
Supplementary Material
Supplementary materials are available at Horticultural Science and Technology website (https://www.hst-j.org).
- HORT_20230052_Table 1s.pdf
Coverage, Simpson, and Shannon Indexes for 16S r RNA Sequencing of the Samples
