

Introduction
Current Status of Genome Sequencing in Vegetable Crops
Application of Genomic Resources
Gene Discovery and Allele Mining
Marker-Assisted Selection (MAS)
Genomic selection
Conclusions
Introduction
Human selection based on phenotype during and after domestication has led to dramatic changes in agronomically important traits (e.g., fruit size) in various crop species. The rediscovery of Mendel’s laws in 1900 established the scientific basis for plant breeding to develop superior plant phenotypes through the creation of genetic variations, selection, and fixation of favorable alleles for human use (Moose and Mumm, 2008). Plant breeding practices have spawned the Green Revolution, which has saved over one billion people from starvation worldwide (Evenson and Gollin, 2003 ). However, the worldwide human population has continually increased and is expected to reach 9.7 billion by 2050 (United Nations, 2015). Recently, climate change has become a serious threat to stable food production (Varshney et al., 2011). To meet the world’s growing demand for food in a fragile environment, a new paradigm of plant breeding is required for crop improvement, which involves integrating genomic resources, high-throughput genotyping and phenotyping technologies, and biotechnology. This paradigm change will accelerate the next green revolution for sustainable food production worldwide.
Selection during conventional plant breeding is based on phenotype instead of genotype. This process is generally laborintensive and time-consuming. In addition, for some traits, phenotypic selection is not very effective due to the difficulty in measuring phenotypes and identifying progenies with the best breeding value (Moose and Mumm, 2008). Conversely, genotyping-based selection via molecular techniques is often a more cost-effective, rapid, precise process than phenotypingbased selection (Yang et al., 2016). Breeding efficiency has increased approximately two-fold using molecular markers relative to phenotypic selection in corn, soybean, and sunflower breeding populations (Eathington et al., 2007). In the past decades, quantitative trait loci (QTL) mapping studies have been extensively conducted to develop molecular markers for markerassisted selection (MAS) in many crop species. Recent advances in next generation sequencing (NGS) technologies have provided high-throughput, cost-effective DNA sequencing methods, opening up opportunities to explore correlations between genetic and phenotypic variation with higher resolution than ever before (Varshney et al., 2014). Using NGS, whole-genome sequencing has been conducted for many crop species (Michael and Jackson, 2013). These genome sequences provide insights into genetic variation through re-sequencing of different varieties (Barabaschi et al., 2012). The huge amounts of information in DNA sequences allow us to identify genome-wide sequence variations such as single nucleotide polymorphisms (SNPs), which facilitate genome-wide association studies (GWAS) for QTL discovery with no limitation on marker availability. These sequences also provide numerous markers for genomic selection, and they accelerate the introduction of new allelic variants in the genomes of cultivated varieties through targeted modification of specific genes and the identification of useful mutations (Barabaschi et al., 2016).
Vegetables are important components of a balanced, healthy diet, representing a rich source of nutrients, vitamins, and minerals. Increasing vegetable consumption provides health benefits by providing functional phytochemicals associated with the prevention of major diseases, such as cancer and cardiovascular disease (Steinmetz and Potter, 1996). Since vegetable crops produce high quantities of food per unit area, they also play a vital role in food security by supplementing the supply of cereal crops. Moreover, vegetable production is more profitable and increases income-generating opportunities relative to most cereal crop production (Weinberger and Lumpkin, 2007). Global vegetable production increased to 1.13 billion tons in 2013 from 516 million tons in 1993 (FAO, 2016). Due to the economic importance of vegetable crops, great efforts in vegetable breeding have been made to improve economically important traits such as disease resistance, abiotic stress tolerance, and quality. In this review, we discuss current trends and future prospects for the application of genomic tools to improve vegetable breeding performance.
Current Status of Genome Sequencing in Vegetable Crops
The genomes of several vegetable crops have been sequenced and assembled using Sanger and/or NGS technologies over the past decade (Table 1). Since tomato is a major vegetable and model plant in the Solanaceae family, the tomato genome sequencing project was initiated by an international consortium in 2004. With the efforts of scientists from 14 countries, two tomato genomes were uncovered using the inbred tomato cultivar ‘Heinz 1706’(Solanum lycopersicum L.) and its closest wild relative, LA1589 (S. pimpinellifolium L.) (The Tomato Genome Consortium, 2012). For the cultivated tomato, 760 megabases (Mb) were assembled into 91 scaffolds across 12 chromosomes, representing 84.4% of the estimated tomato genome (900 Mb). A total of 34,771 protein-coding genes were predicted by the international tomato annotation group (ITAG) v2.3. The availability of the reference genome assembly of ‘Heinz 1706’ has accelerated NGS-based genome sequencing efforts for a large number of tomato varieties (Aflitos et al., 2014; Causse et al., 2013; Jung et al., 2016; Lin et al., 2014). Causse et al. (2013) sequenced the genomes of eight breeding lines (four cherry-type and four large-fruited tomatoes) selected from a collection of 360 varieties based on their molecular diversity. The authors detected more than 4 million SNPs and 128,000 insertion and deletions (InDels) by comparing the eight genome sequences with the reference genome. Aflitos et al. (2014) sequenced a selection of 84 tomato accessions and constructed reference genomes for three wild species, including LA2157 (S. arcanum), LYC4 (S. habrochaites), and LA0716 (S. pennellii). These genome sequences provide insights into tomato genome evolution based on high-confidence sequence polymorphisms. Furthermore, Lin et al. (2014) investigated the molecular signature of human selection in tomato by sequencing the genomes of 360 tomato accessions representing a diverse collection of cultivated varieties and wild species, with an average coverage of 5.7x. Recently, Jung et al. (2016) generated the genome sequences of four elite tomato lines from two private breeding programs in Korea and identified genome-wide SNPs (average of 0.42 per kb) and InDels (average of 0.05 per kb).
Hot pepper, a member of the Solanaceae family, is another economically important vegetable. The reference genome of cultivated pepper (Capsicum annuum L.) was constructed by sequencing Mexican landrace CM334 at 186.6x coverage (Kim et al., 2014). For the reference genome, 3.06 gigabases (Gb) were assembled into 37,989 scaffolds across 12 chromosomes, representing 87.9% of the predicted genome size (3.48 Gb). Pepper genome annotation (PGA) v1.5 predicted a total of 34,903 protein-coding genes, which is similar to that of tomato (34,771 genes based on ITAG v2.3) and potato (39,031 genes based on potato genome sequencing consortium [PGSC] v3.4). In addition to the reference genome, this study generated three additional genome sequences for two pepper cultivars (Perennial and Dempsey) and a wild species, C. chinense (PI 159236), to explore genetic variations and genome structures among varieties. Qin et al. (2014) constructed two new reference genomes using the cultivated pepper line Zunla-1 and the wild progenitor Chiltepin (C. annuum var. glabriusculum). These reference genomes were compared with 20 re-sequencing accessions to explore the molecular footprints of human selection and to identify candidate domestication genes (Qin et al., 2014).
Table 1. Reference genome assembles for vegetable crops in the Solanaceae and Cucurbitaceae families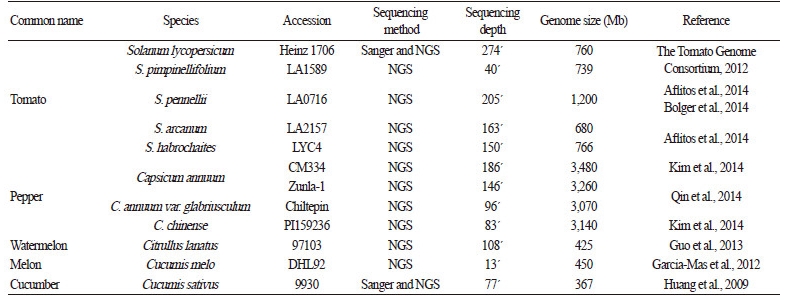 |
Among members of the Cucurbitaceae family, the reference genome of cultivated cucumber (Cucumis sativus L.) was constructed by sequencing Chinese inbred line 9930, which is widely used for modern cucumber breeding (Huang et al., 2009). A total of 26.5 billion bases (72.2x coverage) were generated, and 243.5 Mb were assembled for the cucumber reference genome, which is approximately 30% smaller than the estimated genome sizes of 350–367 Mb. Reannotation of the cucumber genome using RNA-Seq reads predicted 23,248 protein-coding genes, with 25,600 transcripts (Li et al., 2011). A recent study generated 632 Gb of genome sequences by deep re-sequencing of 115 cucumbers, at an average coverage of 18.3x (Qi et al., 2013). This core collection consisted of 102 C. sativus var. sativus and 13 C. sativus var. hardwickii lines, representing 77.2% of the total genetic diversity estimated in a worldwide collection of 3,342 accessions. Analysis of these genome sequences revealed 112 putative domestication sweeps, as well as the genomic basis of divergence among cultivated cucumber populations (Qi et al., 2013).
Watermelon (Citrullus lanatus) is another major crop in the Cucurbitaceae family. NGS-based de novo sequencing of Chinese inbred line 97103 generated a total of 46.18 Gb of high-quality sequences (108.6x coverage) (Guo et al., 2013), 353.5 Mb of which were assembled into 1,793 scaffolds for a draft genome, representing 83.2% of the estimated watermelon genome (~425 Mb). Annotation of the assembled watermelon genome resulted in the prediction of 23,400 high-confidence protein-coding genes, which is close to the 23,248 genes predicted in the cucumber genome (Guo et al., 2013). This study also characterized nucleotide diversity in watermelon germplasm and identified potential selective sweeps by re-sequencing of 20 representative accessions at 5–16x coverage. The re-sequencing panel consisted of 10 cultivated accessions (C. lanatus subsp. vulgaris), six semi-wild accessions (C. lanatus subsp. mucosospermus), and four wild accessions (C. lanatus subsp. lanatus).
For melon (Cucumis melo L.), the genome of double-haploid line DHL92 was constructed by NGS-based whole-genome sequencing (Garcia-Mas et al., 2012). After filtering the mitochondrial and chloroplast genome sequences, both NGS and Sanger sequences (13.5x coverage) of the DHL92 line were assembled into 375 Mb of sequence, representing 83.3% of the estimated 450 Mb melon genome. The melon genome annotation predicted a total of 27,427 genes, with 34,848 transcripts encoding 32,487 polypeptides (Garcia-Mas et al., 2012). Gene prediction suggested that the melon genome has a similar number of genes to that of other Cucurbitaceae crops. Recently, the genetic variation of four melon accessions was investigated by whole genome re-sequencing (Natarajan et al., 2016). This study generated an average of 15.62 million reads per accession, which covered 82.64% of the reference genome assembled by Garcia-Mas et al. (2012). A comparison of sequences of the four accessions to the reference genome revealed 1.53–2.03 million SNPs and 0.42–0.57 million InDels.
Application of Genomic Resources
Gene Discovery and Allele Mining
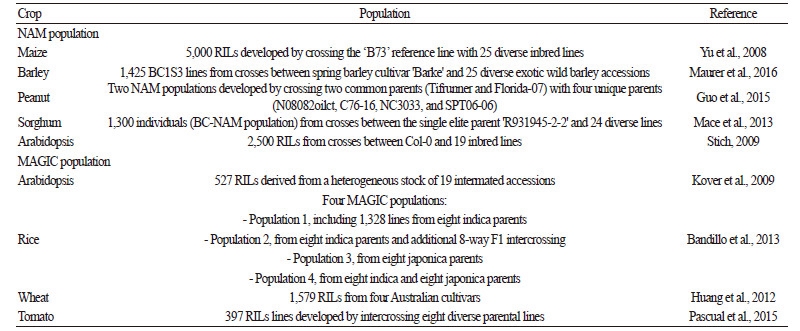
The discovery of favorable genes has been a major goal of vegetable breeding aimed at improving agronomically important traits (Pascual et al., 2015). For nearly two decades, linkage analysis has been extensively conducted to identify QTL using segregating populations derived from biparental crosses (Heffner et al., 2009). F2, backcross (BC), doubled haploid (DH), and recombinant inbred line (RIL) populations are commonly used as biparental mapping populations for linkage analysis (Table 2). Previous linkage analysis studies have identified a number of QTL responsible for traits of interest in vegetable crops,including disease/insect resistance, abiotic stress tolerance, and fruit traits (Foolad, 2007). However, the use of biparental populations provides low mapping resolution due to the occurrence of only a few recombination events (Castro et al., 2012) (Table 2). Thus, QTL detection based on biparental populations is often ineffective for marker-assisted selection using breeding populations where recombination occurs more frequently (Holland, 2007).
Table 2. Comparison of linkage analysis and association mapping approaches for dissecting complex quantitative traits |
Association mapping is a powerful method that uses historical recombination events for QTL detection in natural populations or germplasm collections (Gupta et al., 2005; Zhu et al., 2008). This mapping approach has several advantages relative to linkage analysis, including 1) higher mapping resolution, 2) less time-consuming, and 3) a greater number of alleles to mine (Yu and Buckler, 2006) (Table 2). NGS technologies have provided the opportunity for association mapping by facilitating the development of genome-wide molecular markers, especially SNPs, for high-throughput genotyping. Using a large number of markers, association mapping can be utilized to dissect important complex traits at the genome level via GWAS. In addition to NGS-based marker development, new statistical methods have been developed for association mapping (Price et al., 2006; Pritchard et al., 2000; Yu et al., 2006).
Despite their economic value, progress in association mapping and GWAS of vegetable crops has been slow relative to that for cereal crops. Tomato is a leading vegetable crop for genomic and genetic studies. Several association mapping studies in tomato, including GWAS, have been conducted for fruit traits, volatiles, and disease resistance (Ranc et al., 2012; Shirasawa et al., 2013; Sim et al., 2015; Zhang et al., 2015, 2016). Ranc et al. (2012) developed a core collection of 90 accessions derived from a total of 360 accessions to detect the loci responsible for fruit traits and plant architecture. A total of 352 SNPs and InDels from 81 DNA fragments on chromosome 2 were used to detect associations between markers and fruit traits (fruit weight, locule number, and soluble content). This study validated previously identified candidate genes (fw2.2 for fruit weight, ovate for pearshaped fruit, cnr for non-ripening fruit, and lcn2.1 for locule number) and detected new QTL for fruit traits (Ranc et al., 2012). Shirasawa et al. (2013) identified genome-wide SNPs representing the cultivated tomato genome by re-sequencing six accessions. Of these, 1,293 SNPs were used to genotype 663 accessions for GWAS. A total of nine SNP loci were significantly associated with eight morphological traits, including inflorescence branching, plant habit determinate, plant height, number of leaves between inflorescences, fruit size, locule number, green shoulders on immature fruit, and color of the fruit epidermis (Shirasawa et al., 2013). The loci for tomato volatiles and fruit quality traits were recently investigated via GWAS using 182 SSR markers in a diverse collection of 174 accessions consisting of 123 cherry (S. lycopersicum var. cerasiforme) and 51 heirloom (S. lycopersicum) tomatoes (Zhang et al., 2016, 2015). Association mapping was used to detect previously known genes and novel QTL for bacterial spot resistance in tomato using 461 markers, including 442 SNPs, in a complex breeding population (Sim et al., 2015). Population structure and linkage disequilibrium were investigated in 96 accessions of pepper (C. annuum) via association mapping, representing a wide geographical area worldwide (Nimmakayala et al., 2014a). In addition, this study detected a QTL for both capsaicin and dihydrocapsaicin levels on chromosome 1, as well as three major QTL for fruit weight on chromosomes 8, 9, and 10, respectively. Furthermore, candidate gene association mapping revealed Pun 1 as a key regulator of the production of capsaicinoids and precursors of acyl moieties of capsaicinoids in the capsaicin pathway (Reddy et al., 2014a). Recently, a core collection of 350 accessions was developed from 4,652 worldwide accessions for GWAS in pepper (Lee et al., 2015).
In watermelon, an association mapping study was conducted using 201 SSRs in 96 accessions consisting of 90 sweet watermelons (C. lanatus var. vulgaris) and six egusi types (C. lanatus var. mucosospermus) collected throughout the world (Reddy et al., 2014b). Two QTL on chromosomes 4 and 7 were detected for fruit length over a two-year period. One QTL on chromosome 10 was significantly associated with both fruit length and fruit width, but only in the 2013 season. A marker associated with rind thickness was also found on chromosome 2. In a study by Nimmakayala et al. (2014b), GWAS using 5,254 SNPs in a sweet watermelon collection resulted in the detection of four QTL for soluble solid content. In melon, 87 accessions were collected from wide geographical regions for association mapping (Tomason et al., 2013). This study identified a number of major QTL across 12 chromosomes for five traits: fruit shape (four QTL), fruit length (four QTL), fruit diameter (five QTL), soluble solid content (seven QTL), and rind pressure (two QTL). GWAS was recently conducted for tuberculate fruit and gynoecium traits in cucumber using genome structural variations (SVs) including deletions, insertions, inversions, and tandem duplications (Zhang et al., 2015). The genome-wide SVs were investigated using previous re-sequencing data (Qi et al., 2013) for a core collection of 115 cucumber accessions, and the resulting SVs were used to identify QTL for sex type and fruit traits. Seven QTL were strongly associated with the tuberculate fruit trait. A significant association with gynoecy was found in a 30.2- kb duplicated region that includes the Female (F) locus (Zhang et al., 2015).
Although association mapping has several advantages relative to linkage analysis, rare alleles are not often successfully detected using this approach. The use of multi-parent populations with advanced intercross lines combines the advantages of linkage analysis and association mapping (Pascual et al., 2015; Yu et al., 2008). Two main multi-parent populations, nested association mapping (NAM) and multi-parent advanced generation intercross (MAGIC) populations, have become attractive to breeders (Pascual et al., 2015). These populations require different crossing schemes but similar methods for dissecting complex traits using a large number of markers and progenies from diverse parental founders. The multi-parental populations can be used to generate rich sources of phenotypes by shuffling different alleles in the genome, thus allowing effective QTL discovery using different founders and quick identification of causal variants, as well as rare alleles (Pascual et al., 2016). Furthermore, the NAM and MAGIC approaches provided high statistical power in a cost-effective genome scan performed via computer simulations based on empirical marker data and complex traits with different genetic architecture (McMullen et al., 2009; Yu et al., 2008). However, family-base crossing using multi-parental populations for QTL detection may be affected by genetic background and epitasis (Pascual et al., 2015).
A NAM population was first developed in maize (Yu et al., 2008), followed by several crop species including wheat (Bajgain et al., 2016), barley (Maurer et al., 2016; Nice et al., 2016), peanut (Guo et al., 2015), sorghum (Mace et al., 2013), and Arabidopsis thaliana (Stich, 2009) (Table 3). A MAGIC population was initially described in Arabidopsis (Huang et al., 2011; Kover et al., 2009), followed by rice (Bandillo et al., 2013) and wheat (Huang et al., 2012) (Table 3). However, NAM and MAGIC populations of vegetable crops are not widely used. The first MAGIC population in tomato, consisting of 397 RILs, was developed by intercrossing eight diverse parental lines (Pascual et al., 2015). The re-sequencing of eight founders generated over four million SNPs; of these, a core set of 1,345 SNPs was selected for QTL detection. Haplotype origin was predicted for 89% of the MAGIC line genomes, allowing QTL detection at the haplotype level. Nine QTL, including six location-specific QTL, were mapped for fruit weight in tomato (Pascual et al., 2015). In addition, 63 QTL were detected for eight traits, including truss height, flowering date, fruit weight, firmness, external color, soluble solids content, pH, and titratable acidity, using the MAGIC population (Pascual et al., 2016). The advanced technique of QTL detection using genome-wide markers has accelerated the development of an efficient MAS strategy for vegetable breeding.
Marker-Assisted Selection (MAS)
A number of molecular marker-associated traits of interest have been reported in several vegetable species of the Solanaceae and Cucurbitaceae families (Tables 4 and 5). These markers represent a useful resource for selection during plant breeding. MAS has several advantages relative to phenotype-based selection: 1) MAS is cost-effective and less time-consuming; 2) MAS can be carried out at the seedling stage and in early generations; and 3) genotype-based selection is neutral for environment effects (Collard and Mackill, 2008). MAS, an attractive breeding strategy, has been applied to improve agronomically important traits in vegetable crops, such as disease resistance. Tomato is one of the first vegetable crops for which MAS was applied for various breeding purposes (Foolad and Panthee, 2012). The isozyme marker for acid phosphatase (Aps-11 locus) was first used to select for nematode resistance in a collection of California tomato varieties (Filho and Stevens, 1980). Subsequently, several markers have been employed for backcross breeding to improve resistance to diseases including black mold (Robert et al., 2001), bacterial canker (Coaker and Francis, 2004), and bacterial spot (Rx-3) (Sim et al., 2015; Yang and Francis, 2005). In pepper, MAS has been applied to select breeding lines with resistance against Phytophthora capsici (Thabuis et al., 2004), TSWV (Moury et al., 2000), and bacterial spot (Truong et al., 2011) diseases. Furthermore,‘Maru Salad’, a new pepper cultivar containing high capsinoids levels, was developed using DNA markers for the p-AMT and Pun1 genes (Tanaka et al., 2014).
MAS was effectively utilized to detect watermelon cultivars resistant to Fusarium oxysporum f. sp. niveum (Fon), which causes vascular wilt disease (Lin et al., 2009). In addition, two male-sterile watermelon lines were developed via backcrossing using both the ‘ms’ male sterility and ‘dg’ delayed-green seedling markers (Zhang et al., 1996). Similarly, a melon breeding line with both F. oxysporum-resistance genes and gynoecious traits was successfully selected using molecular markers associated with the sex determination genes ACS7 and WIP1 and the F. oxysporum-resistance gene Fom-2 (Gao et al., 2015). Breeding lines with multiple lateral branching traits were selected from a backcrossing population of cucumber using five associated markers (Fazio et al., 2003; Robbins et al., 2008). In addition, 12 markers were employed to screen F4 and F5 progeny from a cross between two RILs for improved yields and fruit quality (Behera et al., 2008) and to select breeding lines with high-yield, indeterminate, and gynoecious traits (Behera et al., 2010).
Table 4. Molecular markers associated with disease resistance and other traits used for marker-assisted selection in tomato and pepper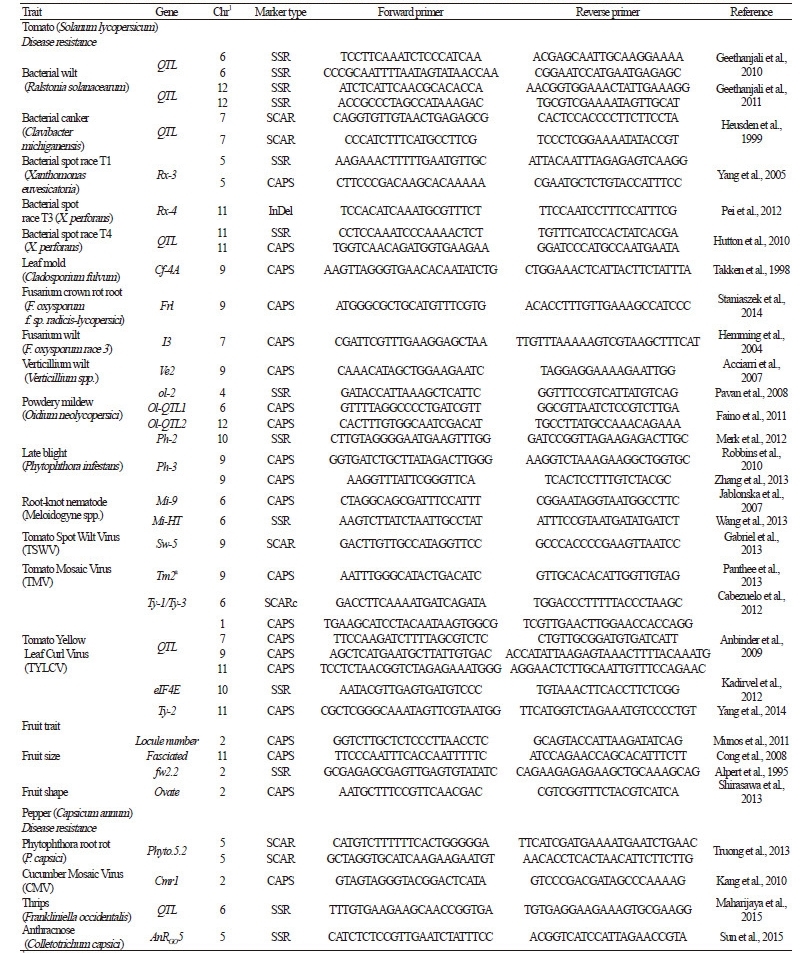 |
Table 5. Molecular markers associated with disease resistance and other traits used for marker-assisted selection in melon, watermelon, and
cucumber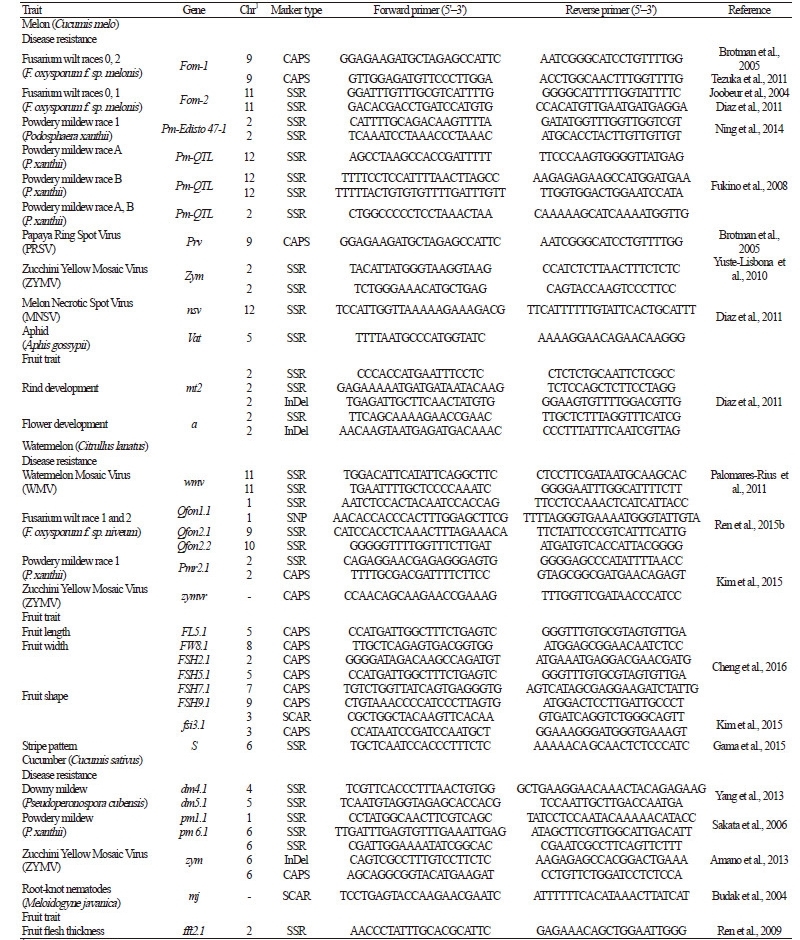 |
Background selection using a number of markers representing a genome facilitates rapid recovery of the recurrent parent genome during backcross breeding. NGS technologies have led to the development of genome-wide markers such as SNPs, SSRs, and InDels in several vegetable crops, including tomato (Sim et al., 2012), pepper (Nicolai et al., 2012), watermelon (Liu et al., 2016; Reddy et al., 2014b; Ren et al., 2015a; Shang et al., 2016), melon (Blanca et al., 2012), and cucumber (Wei et al., 2014; Zhang et al., 2015; Zhu et al., 2016). These marker resources have accelerated background selection during backcross breeding in both the public and private sectors.
Genomic selection
Current MAS methods have been successfully applied to plant breeding, but these methods have limited value for detecting complex quantitative traits underlying numerous small-effect QTL (Dekkers and Hospital, 2002; Xu et al., 2012). Although advances in QTL analysis using genome-wide markers such as GWAS can increase the accuracy and resolution of QTL detection, it is difficult to detect small-effect QTL (Manolio et al., 2009). Moreover, it remains difficult to perform accurate phenotypic evaluation of quantitative traits in large populations (Duangjit et al., 2016). Genomic selection (GS) is an emerging method that bypasses the current limitations of MAS during plant breeding. For GS, genomic estimated breeding values (GEBVs) are calculated by estimating all marker or haplotype effects across the entire genome (Meuwissen et al., 2001). A defined subset of markers associated with traits is currently required for MAS, but all markers are jointly used to predict the breeding values of individuals in a population for GS. Having an appropriate “training” population is key for accurate GEBV calculation and thus, this population must represent the breeding population. Both phenotypic and genotypic data for the training population are used to estimate the parameters of statistical models to calculate GEBVs for individuals using only genotypic data for the breeding population (Meuwissen et al., 2001).
For the past several years, GS has been extensively utilized in cereal crops including wheat (He et al., 2016; Poland et al., 2012), barley (Iwata and Jannink, 2011), and maize (Gorjanc et al., 2016), whereas only a few such studies have been conducted in vegetable crops. Duangjit et al. (2016) investigated the efficiency of GS for a total of 45 metabolomic and quality traits using a collection of 163 tomato accessions, finding that the accuracy of GS was significantly influenced by training population size, marker number/density, and relatedness between individuals (Duangjit et al., 2016). A training population representing 75% of the accessions showed the best accuracy for GEBV prediction. Increasing the number of markers led to stable prediction accuracy, while a marker density of ≥ 0.1 cM distance reduced the accuracy of GEBV prediction. In addition, the more closely related the individuals, the better the accuracy of prediction (Duangjit et al., 2016).
Conclusions
Conventional plant breeding based on phenotypic selection has led to dramatic changes in many agronomic traits (e.g., yield and disease resistance) in various crop species. However, the rapid increase in the worldwide human population and climate change present great challenges for stable food production. NGS technologies have revolutionized genomic research by providing massive parallel sequencing methods. Thus, whole-genome sequencing and re-sequencing are commonly used techniques for crop species. In this genomic era, genome-assisted breeding is emerging as a solution to help meet the great demands for food and nutrients. Vegetable crops provide high quantities of food and represent a rich source of nutrients, vitamins, and minerals for the human diet. Recently, many advances have taken place to facilitate the development of genomic resources for several vegetable crops, including tomato, pepper, cucumber, and watermelon. These genomic resources have facilitated the development of genome-wide markers and high-throughput genotyping platforms. In addition, several mapping approaches have been applied to dissect complex quantitative traits with higher accuracy relative to linkage analysis. These genomic resources and tools are highly valuable for the research community, and they will benefit breeders aiming to develop superior vegetable cultivars with improved traits by increasing the selection efficiency through marker-assisted selection and/ or genomic selection. Moreover, current and future genomic advances will lead to the next green revolution and will facilitate sustainable agriculture.
