

서 언
재료 및 방법
분석 데이터 수집 및 전처리
Read mapping 및 정량화
차등발현 유전자 선발
Gene set enrichment analysis (GSEA)
PoRAS 기반 전사체 분석 파이프라인 구축
결과 및 고찰
PoRAS 파이프라인 구축
고추-감자역병균 상호작용 RNA-seq 분석을 통한 Pre-analysis 검증
유전자 발현 정량화 및 샘플 검증을 통한 Core-analysis 검증
고추-감자역병균 유용 유전자 발굴
고추-감자역병균 상호작용 핵심 기능 분석
서 언
병원균은 작물 생산량과 품질에 영향을 끼치는 주 요인 중 하나로써 전 세계적으로 작물의 약 30%이상이 병원균에 의해 손실된다고 알려져 있다(Chun et al., 2011). 인류는 병원균의 피해를 최소화하기 위하여 화학적 방제법을 주로 사용해왔지만, 지속적인 농약사용은 토양의 산성화 및 잔류독성, 환경오염 등의 문제를 일으켜 왔다(Bitew and Alemayehu, 2017). 이러한 문제에도 불구하고 현재까지도 병원균에 의한 피해를 방제하기 위해 화학적 방제를 사용하고 있으며 현재의 상황을 개선하기 위해서는 저항성 형질을 가진 유전자원을 활용한 병저항성 품종 육성을 통한 자연 친화적 방제가 중요하다(Martin, 2011; Kwon et al., 2021; Kang et al., 2022a).
감자역병은 Phytophthora infestans라는 난균류에 의해 발병되며, 매년 전 세계적으로 약 50억 달러에 이르는 피해를 입힌다고 알려져 있다(Tsedaley, 2014). 고추의 경우 고추-감자역병 상호작용 스크리닝 분석(Lee et al., 2014) 을 통해 감자, 토마토와는 달리 감자역병균에 대해 비기주 저항성을 가지고 있는 것으로 알려져 있다. 고추-감자역병 상호작용 전사체의 분석의 경우 특정 형질 및 한정된 유전자들을 대상으로 분석되었다(Lee et al., 2017). 따라서 비기주 저항성의 핵심 인자 동정을 위해 전체 유전자에 대한 프로파일링 및 유전자의 기능 연구가 추가적으로 필요하다. 최근에는 다양한 전략을 통해 기주 및 비기주 저항성 유전자를 발굴하고 이를 활용한 새로운 저항성 형질을 발굴하고 육종에 적용하고자 하는 연구가 진행되고 있다(Lee et al., 2016; Seo et al., 2016; Kim et al., 2017; Oh and Choi, 2022).
NGS(Next generation sequencing)기술이 발달함에 따라 유전체 및 전사체 분석 연구가 증가하는 추세이며 이를 통해 식물내 전체 유전자의 변이 및 발현정도를 확인하는 다양한 분석방법이 제안되고 있다. 이러한 분석방법은 인간, 동물, 미생물뿐만 아니라 식물에서도 생명현상을 이해하기위해 다양한 분석에 수행되고 있으며(Conesa et al., 2016; Kim et al., 2020; Lee et al., 2021), 특히 전사체 분석에 사용되고 있는 RNA-seq 방법의 경우 다양한 환경스트레스와 관련된 기작을 규명하기 위해 중요 작물들의 전체 유전자들에서 형질 반응 및 특이성을 가지는 유전자들을 선발함으로써 다양하고 복잡한 저항성 체계를 확인하는데 사용되고 있다(Wang et al., 2009; Lee et al., 2019; Kang et al., 2020). 하지만, RNA-seq을 이용한 전사체 분석은 다양한 분석 프로그램이 제시됨에 따라 다양한 조건을 고려해야 하며 획일화된 분석 방법은 확립되지 않았다(Dobin et al., 2013; Bolger et al., 2014; Fumagalli et al., 2014; Kim et al., 2015).
본 연구에서는 전사체 분석을 위한 기초 전략도를 제시하고 고품질 서열을 확보하여 감자역병균에 반응하는 고추 유전자 전체의 발현패턴을 확보하였다. 또한, 특이적으로 반응하는 고추 유전자 2,594개의 유전자를 선발하여 접종 후 시간대에 따라 반응하는 유전자의 Gene Ontology(GO)를 확인하여 defense response(GO: 0006952)와 같은 저항성 관련 유전자 군집의 반응을 확인하였다. 본 연구를 통해 감자역병균에 반응하는 전체 고추 유전자의 반응성을 확인하였고, 그에 따라 제시된 전략을 통해 RNA-seq 데이터를 기반으로 한 분석을 통해 핵심 후보 인자를 선발에 유용하게 사용될 수 있음을 확인하였다.
재료 및 방법
분석 데이터 수집 및 전처리
본 연구는 고추(Capsicum annuum ‘CM334’)의 잎에 감자역병균을 처리하여 각 7개의 시간대(0h, 6h, 12h, 1D, 2D, 3.5D, 5D)의 반응을 확인하기 위한 RNA-seq 데이터를 사용하였다. 해당 데이터는 NCBI의 Sequence Read Archive database에 공개된 SRP106410(Kim et al., 2018) 데이터를 이용하여 수행하였다. 각 처리는 3반복으로 샘플링 되었으며 Hiseq 2500을 통해 sequencing되었다.
전체 RNA-seq 데이터에서 어댑터 서열 및 부정확한 서열의 제거는 각각 Cutadapt(Martin, 2011) 와 Trimmomatic(Bolger et al., 2014) 프로그램을 이용하여 수행하였다. Filtering의 기준은 Phred score < 20을 기준으로 제거하였으며 추가로 단일 서열의 길이가 36bp이하인 서열은 제거하였다(Lee et al., 2020; Kim et al., 2021). 이후 각 sequencing 데이터의 검증을 위해 FastQC(Andrews, 2010) 와 MultiQC(Ewels et al., 2016) 프로그램으로 데이터의 품질을 확인하였다.
Read mapping 및 정량화
전처리를 통해 확보한 clean reads는 HiSat2(Kim et al., 2015) 프로그램을 통해 고추 유전자(Annuum.v.2.0.CDS.fa)에 mapping시켰다. Mapping에 사용한 고추 유전자 정보는 “Pepper Genome Database”(http://peppergenome.snu.ac.kr/)에서 확보하였다. 고추 유전자 서열에 정렬된 read는 FPKM(fragments per kilobase of transcript per million)으로 발현 값을 보정하였으며(Ding et al., 2015) in-house script를 통해 계산하였다. 유전자의 발현을 통해 sample간 차이를 검증하기 위해 주성분분석(principle component analysis, PCA) 및 sample clustering분석을 수행하였고, 각 분석 및 시각화는 R studio프로그램을 통해 수행되었다.
차등발현 유전자 선발
정량화를 통해 확보한 유전자의 발현을 기반으로 대조군과 실험군의 차이를 확인하기 위해 DESeq2 bioconductor package(Love et al., 2014)를 통해 분석을 수행했다. 각 시간대(6h, 12h, 1D, 2D, 3.5D, 5D)에 따라 control(TDW 접종) 대비 발현이 4배 이상(Log2FC > |2|) 차이를 보이고 FDR값이 0.05이하인 유전자를 기준으로 DEG(differentially expressed gene)를 선발하였다. 각 DEG에 따른 발현의 시각화는 R studio프로그램을 통해 수행되었다. boxplot과 heatmap은 R studio의 boxplot함수와 ComplexHeatmap R 패키지를 사용했다(Kang et al., 2018; Kim et al., 2019; Kang et al., 2021).
Gene set enrichment analysis (GSEA)
앞선 분석을 통해 선발된 DEG를 기반으로 각 시간대에 따라 반응하는 유전자들의 기능을 확인하기 위해 GSEA 분석 중 GO 분석을 수행하였다. GO는 R 패키지의 goseq(Young et al., 2012)을 통해 분석을 수행하였으며 FDR < 0.05를 기준으로 filtering 하였다. 전체 GO 결과 중 유전자 군집의 기능을 확인하기 위해 Biological Process(BP)를 중심으로 분석을 수행하였다.
PoRAS 기반 전사체 분석 파이프라인 구축
전사체 분석을 위한 PoRAS 플랫폼은 아래와 같이 2단계로 나누어 각 분석 단계별 세부 파이프라인 생물정보 분석 코드 및 필요 프로그램은 github(https://github.com/Lee-June-Sung/PoRAS)에서 사용 가능하도록 구축하였다. 1단계는 sequencing 이후 불필요한 정보 및 정확한 결과의 확보를 위한 filtering 과정이며 preprocessing 및 QC단계가 포함되어 있다. 각 단계는 Cutadapt, Trimmomatic, FastQC 그리고 MultiQC를 통해 분석을 수행한다. 2단계는 전처리 이후 모든 분석 과정이 포함되며 핵심 유전자의 선발을 위해 구축되었다. 각 단계는 Hisat2, DESeq2 그리고 GOseq 프로그램이 활용되며, R 과 perl 코드를 기반으로 데이터 분석 및 검증을 을 수행하였다.
결과 및 고찰
PoRAS 파이프라인 구축
RNA-seq 분석은 크게 Pre-analysis, Core-analysis 및 Advanced-analysis로 분류하여 분석을 수행하며 이는 대부분의 연구에서 공통적으로 적용되고 있다(Conesa et al., 2016). 본 연구에서는 Pre-analysis와 Core-analysis의 일부 분석을 위한 파이프라인을 구축하여 샘플에 대한 모든 유전자의 발현패턴을 확인하고 핵심 유전인자의 발굴을 위한 분석을 수행하는 기초 파이프라인을 구축하였다(Fig. 1). 먼저 Pre-analysis에 해당하는 Pre-processing 단계에서는 raw data의 품질 향상을 위해 전처리 과정을 수행한다. 이 때, Cutadapt를 통해 sequencing 과정에서 생성되는 adaptor 서열을 제거하여 임의로 생성된 서열을 제거한다. 이후 Trimmomatic을 통한 sequencing 서열 정확도 보정을 수행하여 개별 read에서 정확도가 떨어지는 서열을 제거함으로써 sequencing 과정에서 생성되는 에러를 최소화한다. 이 두 과정을 통해 생성된 RNA-seq data는 adaptor 서열이 없는 고품질 데이터로 이후 core-analysis 단계에서 에러를 최소화하여 정확한 정보를 확보할 수 있다.
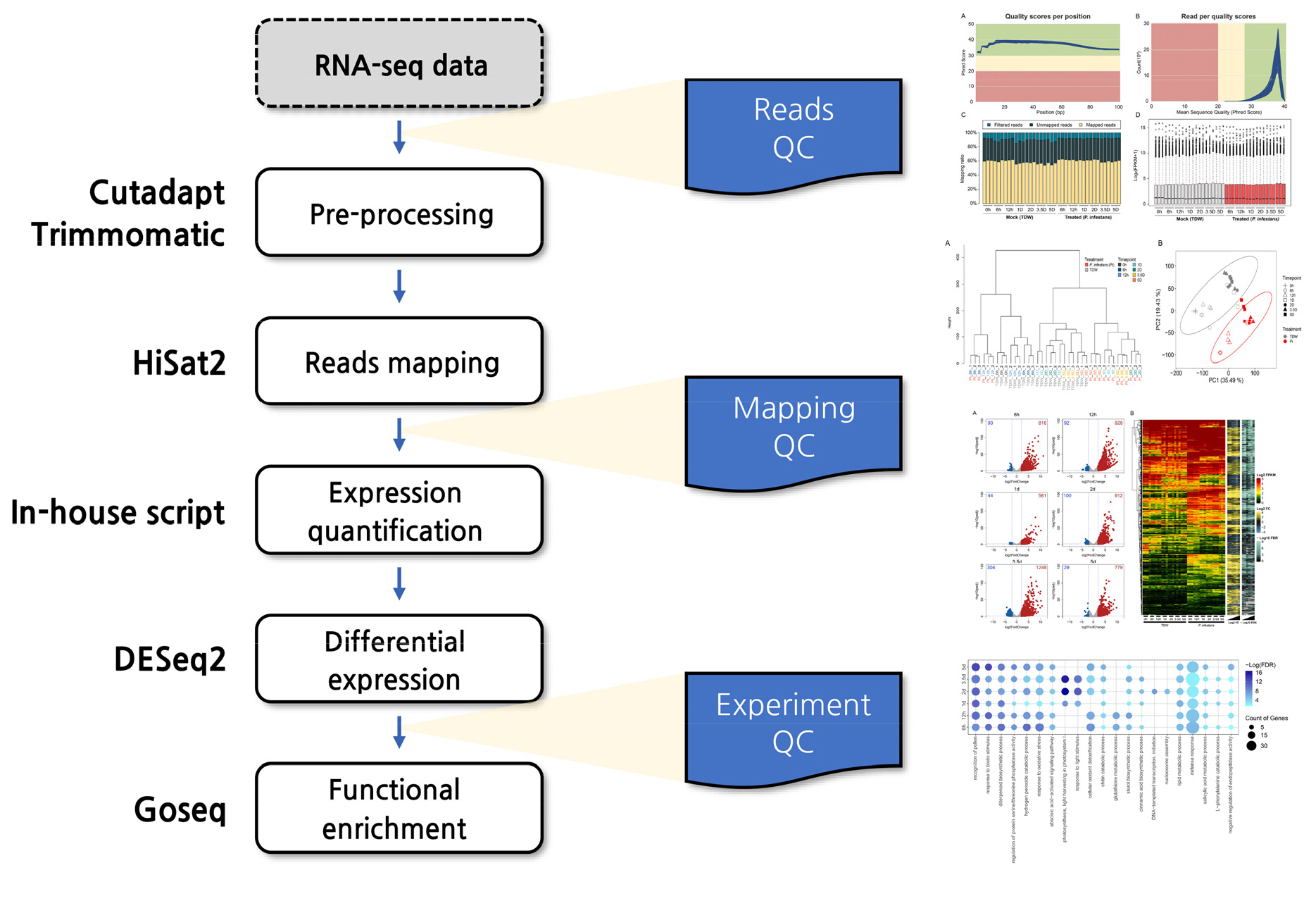
Core-analysis에 해당하는 Read mapping ~ Functional enrichment 과정은 전처리 이후의 고품질 데이터를 기반으로 유전자 전체에 대한 발현패턴을 확보하고 핵심 유전인자를 선발하는 과정이다. 먼저 HiSat2를 이용한 Read mapping을 통해 유전자 서열과 유사한 read를 정렬한다. 이후 mapping 결과를 수치화하여 유전자들에 대한 발현을 확인하며 본 연구에서는 FPKM으로 유전자들의 발현을 정량화하였다. 이를 통해 각각의 유전자에 대한 발현을 확인할 수 있지만 효과적으로 비교하기 위해 통계적으로 유의한 차이를 나타내는 유전자들을 선발하는 과정을 거친다. 해당 과정은 DESeq2를 통해 유전자의 발현을 비교하며 본 과정을 통해 대조구(여기서부터 TDW)과 감자역병균 접종 처리구(여기서부터 Pi)간의 발현차이가 유의하게 나타나는 유전자만을 선발할 수 있다. 유전자의 기능이 할당된 reference genome의 경우에는 확보한 DEG의 기능을 보다 쉽게 확인하여 핵심 유전인자를 확인할 수 있지만 기능에 대한 유전자 군집의 확인하기 어려운 유전체의 경우에는 Functional enrichment와 같은 기능분석을 통해 핵심인자를 선발한다. 본 파이프라인에서는 gene ontology term(GO term)을 통해 DEG의 기능을 확인할 수 있도록 구축하였으며 유의성 기준을 통과한 GO term을 통해 유전자 군집의 기능을 유추하여 핵심 인자를 선발할 수 있다.
각 단계별 분석을 수행할 때 오류를 최소화 하기위해 각 단계별 Quality check(QC) 과정을 추가하여 정확도를 보정하였다. Pre-analysis 과정에서 확보되는 fastq 파일의 점검을 위해 FastQC와 MultiQC를 통해 각 데이터의 sequencing error와 정확도를 확인하여 정상적인 데이터를 선별할 수 있다. 이후 유전자의 발현을 수치화한 후 Principal component analysis(PCA)분석 및 sample clustering을 통해 데이터간 variation을 확인하여 동일 처리 샘플간 또는 Pi와 TDW간의 차이를 확인하여 데이터의 오염과 같은 데이터 생성간 발생할 수 있는 오류를 줄일 수 있도록 구축하였다. 마지막으로 DEG 분석 및 Functional enrichment 과정을 통해 각 데이터가 유추가능한 유전자의 반응성을 확인함으로써 Pi 처리가 정상적으로 수행되었는지 점검 가능하도록 설계하여 각 분석과정 및 결과의 신뢰도와 정확도를 높일 수 있는 파이프라인을 구축하였다.
고추-감자역병균 상호작용 RNA-seq 분석을 통한 Pre-analysis 검증
파이프라인의 검증을 위해 고추-감자역병균 상호작용 RNA-seq 데이터를 사용하였다. 데이터는 최소 raw read가 8,671,868 최다 raw read는 17,324,392로 확인하였다(Table 1 and Suppl. Table 1). 이후 전처리 과정을 수행하였고, 어댑터 서열 및 저품질 서열을 제거함으로써 전체 데이터에서 고품질의 정보만을 확보하였다. 이를 통해 확인된 clean read는 최소 8,005,004 최다 15,757,564로 확인되었으며 84.86–92.37%의 고품질 서열이 남은 것을 확인하였다(Table 1 and Suppl. Table 1). 이후 해당 서열들의 품질을 세부적으로 확인하고자 FastQC 및 MultiQC 프로그램을 통해 전사체 데이터들의 품질을 확인하였다. 전체 샘플에서 위치에 따른 reads들의 서열 위치 기반 quality score를 확인한 결과 read들의 평균 품질은 quality score가 30 이상(base call 정확도 > 99.9%)이 되는 것을 확인하였다(Fig. 2A). 또한, 39개의 모든 샘플들의 샘플 내 전체 서열의 개수기반 quality score를 확인한 결과 모든 샘플에서 20 이상(base call 정확도 > 99%)이 되는 것을 확인하였다(Fig. 2B). 전처리를 통해 확보한 clean read는 raw data 대비 평균 9.36%가 filtering 되었으며(Fig. 2C), 2.0버전의 고추 CDS에 mapping된 비율은 평균 65.52%로 동일 참조유전자서열을 사용한 다른 선행논문(Zhang et al., 2016)의 mapping 비율(53.78–56.38%)보다 더 높은 수치를 보였다. 따라서 본 연구에서 제시한 전처리 방법에 따른 quality control에는 이상이 없는 것으로 확인되었으며 본 분석을 통해 생성된 clean read를 이용하여 이후 Core-analysis에 사용하였다.
Table 1.
Summary Statistics of Pepper-P. infestans Interacted with RNA-seq for the PoRAS-based Transcriptome Analysis
| Samplez | Raw reads | Clean reads | Filtered ratio (%) | Mapped reads | Mapping ratio (%) |
| CM-pi-0 | 12,497,044 | 11,481,985 | 91.88 | 7,558,895 | 65.79 |
| CM-TDW-6h | 14,159,873 | 12,567,949 | 88.57 | 8,515,183 | 67.75 |
| CM-TDW-12h | 14,211,393 | 12,984,145 | 91.41 | 8,674,523 | 66.84 |
| CM-TDW-1D | 10,622,809 | 9,213,476 | 86.81 | 6,025,402 | 65.35 |
| CM-TDW-2D | 12,524,443 | 11,308,969 | 90.33 | 7,273,575 | 64.40 |
| CM-TDW-3.5D | 10,755,521 | 9,618,353 | 89.45 | 5,974,739 | 62.00 |
| CM-TDW-5D | 11,534,617 | 10,177,878 | 88.12 | 6,503,032 | 63.92 |
| CM-pi-6h | 12,016,665 | 11,045,365 | 91.90 | 7,446,744 | 67.38 |
| CM-pi-12h | 12,645,779 | 11,615,109 | 91.86 | 7,718,453 | 66.41 |
| CM-pi-1D | 11,050,668 | 10,138,304 | 91.84 | 6,723,946 | 66.32 |
| CM-pi-2D | 13,870,755 | 12,769,601 | 92.06 | 8,533,244 | 66.81 |
| CM-pi-3.5D | 13,544,129 | 12,445,278 | 91.89 | 7,967,957 | 64.10 |
| CM-pi-5D | 13,423,639 | 12,365,602 | 92.13 | 7,984,655 | 64.63 |

Fig. 2.
Quality assessment of pepper transcriptomes. The filtered reads from all 39 samples were assessed by MultiQC. (A) Mean quality score distribution in each position, (B) Read counts distribution for the mean sequence quality, (C) Filtering and read mapping ratio, (D) The distribution of normalized FPKM for 34,899 pepper gene expressions measured in each sample was confirmed. Data were converted to log2 to prevent values from shifting to one side and to reduce deviations between expression values.
유전자 발현 정량화 및 샘플 검증을 통한 Core-analysis 검증
앞선 과정에서 생성된 유전자의 read count는 샘플 간 유전자의 발현을 비교하기 위해 FPKM으로 정규화하였다. 샘플 내 전체 유전자 발현 값을 확인한 결과 최소 0에서부터 최대 68264.23(log2 FPKM = 16.05)까지 다양하게 분포한다는 것을 확인할 수 있었다(Fig. 2D). 이후 실험군의 각 sample 처리간 오류를 확인하기위해 전체 유전자의 발현패턴을 통해 PCA분석 및 sample clustering을 수행하였다. P. infestans 및 TDW를 접종한 고추 전사체의 시간대별 발현 값 변화를 확인하였고, 대부분의 처리에서 3반복간 동일한 그룹으로 분류되는 것을 확인하였다(Fig. 3). Sample clustering에서 TDW 1d와 TDW 2d 샘플을 제외한 모든 샘플에서 3반복 샘플이 같은 그룹으로 묶이는 것을 확인하였으며 이는 각 반복이 매우 유사한 유전자 발현 패턴을 나타내는 것을 의미한다(Fig. 3A). 또한 PCA분석을 통해 확인한 결과 Pi와 TDW 사이의 유전자들의 발현패턴이 확연히 두 그룹(붉은색-Pi; 회색-TDW)으로 구분되었으며 3반복간 매우 유사한지점에서 확인되는 것을 확인할 수 있다(Fig. 3B). 이는 두 처리가 명확히 다른 유전자 발현패턴을 나타내는 것을 의미하며 3반복의 처리가 정상적인 것을 의미한다. 이를 통해 본 과정을 통해 새로 생산한 데이터 및 오픈소스 데이터를 이용한 데이터에서 sample간 차이를 확인할 수 있으며 이를 통해 처리된 샘플의 특성을 확인하여 본 분석에서의 오류율을 최소화할 수 있다.

Fig. 3.
Sample clustering and principal component analysis of samples with gene expression for validation in the PoRAS QC step. (A) Classification of RNA-seq samples through a hierarchical clustering analysis. Each color represents a certain time point and treatment group, (B) A principal component analysis (PCA) was conducted to determine the degree of variation of each sample, and samples were grouped according to each treatment (P. infestans, TDW). The red group was treated with P. infestans as a treatment and the gray group was treated with triple-distilled water (TDW) as a control.
고추-감자역병균 유용 유전자 발굴
특이적으로 발현된 유전자(DEG)의 선별은 TDW와 비교하여 P. infestans를 접종했을 때 발현 값이 4배 이상 차이가 나는(log2 FC > |2|) 유전자들을 각 시간대(6h, 12h, 1D, 2D, 3.5D, 5D)별로 추출하였다. 그 결과 선발된 2,594개의 DEG에 대한 발현차이를 확인하기 위해 log2 FPKM값을 기반으로 발현패턴을 확인하였다(Fig. 4). 먼저 각 시간대별 비교를 위해 발현 변화를 중심으로 패턴을 확인하였으며 1d에서 DEG가 605개로 가장 적게 나타났으며 3.5d에서 유전자가 1,552개로 가장 많이 변화하는 것을 확인하였다(Fig. 4A). 또한 모든 시간대에서 up-regulation되는 유전자가 down-regulation되는 유전자보다 더 많이 확인된 것을 통해 Pi가 처리됨에 따라 발현이 억제되는 유전자보다 더 강하게 발현이 유도되는 유전자가 많은 것을 의미하며 발현이 변화한 유전자들은 TDW 보다 4배이상 강하게 또는 약하게 발현되는 것으로 식물의 발현변화가 매우 강하게 일어나는 것을 알 수 있다. 이러한 유전자의 발현을 heatmap을 통해 패턴을 확인할 수 있는데 이를 통해 두 샘플간 유전자의 발현변화의 시간대 별 차이를 확인할 수 있다(Fig. 4B). 해당 결과에서 발현된 DEG들은 대부분 TDW대비 Pi처리에서 발현이 강하게 유지되었으며, 이 방법을 통해 확인된 DEG는 선행연구에서 비기주저항성을 유도한다고 제시되었던 CA12g05030(CA.PGAv.1.6.scaffold459.13와 베스트 매치) 와 CA12g05140(CA.PGAv.1.6.scaffold135.6와 베스트 매치)등의 핵심 저항성 유전자들을 포함하고 있다(Suppl. Table 3; Lee et al., 2017). 이를 통해 본 분석방법을 통한 유전자의 발현패턴 확인 및 후보 유전자 선발에 유용하게 사용할 수 있음을 확인하였다.

Fig. 4.
Differentially expressed genes by P. infestans inoculation belonging to the core-analysis stage of PoRAS. (A) Scatter plot based on gene expression changes, and (B) corresponding heatmap. Differential expression genes were selected based on genes expressed two times more or less than the control. The FPKM values and expression change value (log2 FC and -log FDR) for 2,594 genes differentially expressed at all times (6h, 12h, 1D, 2D, 3.5D, and 5D) were log2 transformed and then expressed as heatmaps.
고추-감자역병균 상호작용 핵심 기능 분석
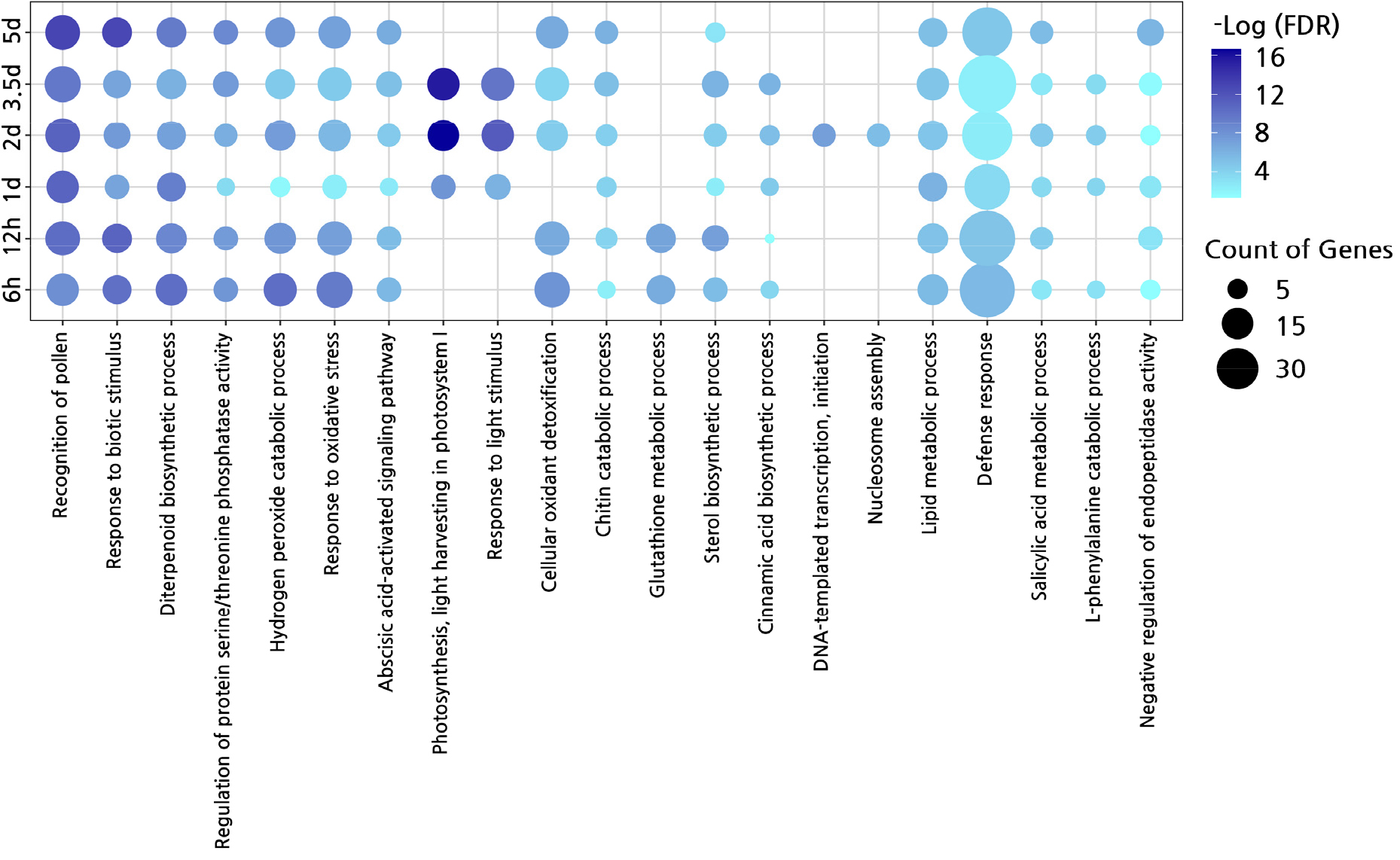
각 시간대에 따른 DEG를 중심으로 유전자군집을 설정하여 GSEA 분석을 통해 유전자 군집의 기능을 확인하였다(Fig. 5). 각 시간대별로 확인된 유전자 군집의 기능은 대부분 유사한 기능을 나타냈고, 공통적으로 defense response에 가장 많은 유전자들의 기능이 확인되었다(Suppl. Table 2). defense response에 해당하는 유전자는 방어반응의 조절인자(GO:0031347; Defensive response regulation), 타 종에 의한 방어기작(GO:0098542; defense response to other organism), 방어반응 부정적인 규제(GO:0031348; negative regulation of defense response) 등의 기능을 가진 유전자들을 포함하고 있고, 병원균에 대한 저항성 반응을 포함한 다양한 외부 환경스트레스 상호작용에서 방어기작 혹은 관련 신호전달에 관여한다고 보고되었다(Lee and Yeom, 2015; Kang et al., 2022b). 이들 GO term을 가진 유전자들은 Pi에 대한 외부 환경 스트레스들에 대한 저항성 반응을 나타내는 핵심 인자로써 기능을 할 것으로 해석할 수 있다. 또한 response of biology stimulus(GO:0050896) 및 recognition of pollen(GO:0048544)이 가장 유의한 차이를 보였다. 해당 결과 중 recognition of pollen(GO:0048544)의 경우에는 시간대가 후기로 넘어갈수록 TDW에 비해 유의성이 높아진다. 이들 그룹에는 자가면역(autoimmunity) 및 신호전달 반응에 관여하는 RLP/RLK를 포함한 리간드 인지 단백질들이 대표적으로 포함된다(Sanabria et al., 2010; Kang and Yeom, 2018; Wan et al., 2021). 특이적으로 2d 와 3.5d에서는 photosynthesis(GO:0015979) 가 가장 유의하게 나타났으며 이 그룹들의 유전자들은 식물 호르몬 반응 조절 및 이를 통해 다양한 스트레스 반응 신호전달에 관여하는 것으로 보고되었다(Attaran et al., 2014; Kang et al., 2020). 이러한 결과를 통해, 감자역병균과의 상호작용 동안 외부 자극에 반응하는 인지 단백질군과 신호전달 관련 유전자군들의 상호작용을 통해 고추에서 감자역병균 대한 비기주 저항성 반응에 관여하는 것으로 예측된다(Lee et al., 2017). 본 분석을 통해 DEG를 기반으로 유전자군집의 기능을 확인할 수 있으며 핵심 기능을 중심으로 저항성 유전인자 혹은 생식 생장관련 유전자를 선발하여 원하는 형질의 유전자를 발굴하는데 유용하게 사용할 수 있다. 따라서 본 연구에서 제시한 전략은 특정 처리에 따라 반응하는 유전자를 선발할 수 있으며 이를 통해 저항성 유전자의 발굴 및 유전자의 기능을 유추하는데 정상적으로 사용할 수 있음을 확인하였다.
Supplementary Material
Supplementary materials are available at Horticultural Science and Technology website (https://www.hst-j.org).
- HORT_20230010_Table_1s.xlsx
Total RNA-seq statistics
- HORT_20230010_Table_2s.xlsx
Gene set enrichment analysis result using PoRAS
- HORT_20230010_Table_3s.xlsx
Gene expression patterns of important resistance genes in the previous study
