

서 언
재료 및 방법
NAC 유전자 동정
NAC 유전자의 모티프 및 서브도메인 구조 분석
Gene Ontology 분석
NAC 유전자의 아미노산 서열 구성 분석
NAC 유전자의 계통학적 분석
NAC 유전자의 복제 분석
저온 스트레스 처리 시 배의 NAC 유전자 발현 분석
결과 및 고찰
NAC유전자 정보 업데이트
NAC 도메인 및 모티프 구성
NAC 유전자들 간의 계통학적 분석
저온 처리 시 유전자 발현 분석
서 언
NAC 전사인자는 식물 특이적인 전사인자로서 식물의 기관 발달과 다양한 생리 및 생물학적 반응에 중요한 역할을 한다(Nakashima et al. 2012). NAC라는 명칭은 NAM(No Apical Meristem), ATAF1/2(Arabidopsis Transcription Activation Factor 1/2), CUC2(Cup-shaped Cotyledon 2)에서 공통적으로 발견되는 보존된 도메인 서열에 기반하여 유래하였으며, 일반적으로 N말단에 보존된 도메인과 C말단에 다양한 기능을 수행하는 가변 영역으로 구성된다(Nuruzzaman et al. 2013). NAC 도메인은 여러 개의 β-sheet로 이루어진 역평행 구조를 가지며, A, C, D 서브도메인은 높은 보존성을, B와 E는 상대적으로 낮은 보존성을 보인다(Kikuchi et al. 2000; Ooka et al. 2003). NAC 전사인자는 세포 분열, 분화, 노화 등 발달 과정뿐만 아니라, 생물학적 및 비생물학적 스트레스 조건에서도 유전자 발현을 조절하여 식물의 스트레스 반응과 환경 적응에 관여하는 것으로 알려져 있다(Nuruzzaman et al. 2013; Diao et al. 2020).
사과(Malus domestica)와 배(Pyrus pyrifolia)는 장미과에 속하는 대표적인 낙엽성 과수로, 전 세계 사과 생산량은 97,339,338톤에 달하며 이 중 60.1%가 아시아에서 생산되는 경제적 및 생태학적으로 중요한 작물이다(FAOSTAT 2023). 사과는 polyphenols, hydroxycinnamic acid, glycosylated flavonoids, vitamin 등 다양한 생리활성 물질을 함유하고 있으며, 항산화 및 항염 효과가 보고되었다(Can et al. 2014; Padua et al. 2014). 배는 carotenoid, 아미노산, 유기산, 섬유질, polyphenols, flavonoids, anthocyanin 등 다양한 영양소를 포함하고 있어 온대 지역에서 중요한 과수 자원으로 활용되고 있다(Li et al. 2016b). 그러나 2023년 국내 사과와 배의 생산량은 전년 대비 각각 30.3%, 26.8% 감소하였으며, 이는 개화기 저온 스트레스로 인한 꽃눈의 흑변 및 고사에 따른 착과율 저하가 주요 원인으로 지적되고 있다(KREI 2024). 이러한 재배 불안정성과 기후 스트레스 대응의 필요성으로 인해 사과와 배의 유전체 기반 연구는 중요성이 강조되고 있으나 NAC 전사인자에 대한 기존 연구는 주로 벼와 애기장대 같은 모델 식물에 국한되어 왔으며, 장미과 과수에서의 연구는 여전히 미흡한 실정이다(Ooka et al. 2003).
본 연구에서는 벼, 애기장대, 사과, 배의 NAC 전사인자를 재동정하고 비교 분석함으로써, 장미과 과수 특이적 진화적 확장과 기능적 의미를 규명하고자 하였다. 이를 위해 모티프와 유전자 구조 분석을 통해 보존 서열을 확인하고, 계통 분석과 CAFE 분석을 통해 사과와 배의 NAC 유전자군의 진화적 특성을 조사하였다. 또한 RNA-seq 기반 발현 분석과 공동 발현 네트워크 분석을 통해 배에서 저온 반응성과 관련된 주요 NAC 유전자를 확인하였다. 본 연구는 NAC 유전자의 구조와 기능에 대한 이해를 확장하고, 장미과 과수의 저온 스트레스 대응 유전자 탐색 및 향후 기능 분석과 분자 육종 연구에 기초 자료를 제공한다.
재료 및 방법
NAC 유전자 동정
본 연구에서는 Oryza sativa ssp. indica, Arabidopsis thaliana, Malus domestica, Pyrus pyrifolia의 유전체 데이터를 이용하여 NAC 유전자군을 재동정하였다. 사용한 4종의 genome data와 그 출처는 Supplementary Table 1에 정리하였으며, 재동정은 TGFam-Finder v1.20(Kim et al. 2020)을 통해 수행하였다. 도메인 식별을 위해 InterProScan 5(-f tsv-appl Pfam) (Jones et al. 2014)로 생성한 TSV 파일을 사용하였고, “TARGET_DOMAIN_ID”는 Pfam database를 기준으로 NAM 도메인에 해당하는 PF02365로 설정하였다. 이때 사용된 매개 변수는 “EXTENSION_LENGTH” = 100,000, “MAX_INTRON_ LENGTH” = 100,000, “HMM_CUTOFF” = 1e-3이었다. 재동정된 NAC유전자에는 기존 동정에 사용된 locus name 대신 새로운 유전자 이름을 부여하였다.
NAC 유전자의 모티프 및 서브도메인 구조 분석
NAC 유전자의 구조적 특성을 규명하기 위해 전체 유전자 서열을 대상으로 MEME v5.1.1(-mod zoops -nmotifs 50 -minw 10 -maxw 50 -objfun se -markov_order 0) (Bailey et al. 2006) 분석을 수행하여 보존된 모티프를 탐색하였다. 이후, 각 모티프의 위치를 확인하기 위해 MAST v5.1.1(Bailey and Gribskov 1998)을 활용하였다. 탐색된 모티프들 중 기존 연구에서 보고된(Ooka et al. 2003) NAC 도메인의 서브도메인 영역에 위치하는 모티프들을 대응시켜, 서브도메인 내에서의 보존된 모티프 구성을 확인하였다. 이를 통해 NAC 유전자의 도메인 구조와 모티프 보존 양상을 종합적으로 분석하였다.
Gene Ontology 분석
NAC 유전자의 기능을 추정하기 위해 OmicsBox v3.4(https://www.biobam.com/omicsbox/)를 이용하여 Gene Ontology(GO) 분석을 수행하였다. NAC 유전자의 아미노산 서열을 BLASTP를 통해 non-redundant proteins database(nr v5)와 정렬하였으며, e-value < 1e-3로 설정하였다. BLASTP 매개변수는 기본 값(Number of hits: 20, HSP length cutoff: 33, HSP hit coverage: 0)을 사용하였다. 이후 InterProScan 5(-f xml-appl Pfam) (Jones et al. 2014)를 통해 생성한 XML 파일을 BLAST 결과와 병합하여, GO term으로 매핑하고 추가적인 기능 동정을 수행하였다. 각 NAC 단백질에 대한 GO term은 biological process, molecular function, cellular component의 세 항목으로 분류하였고, 각 항목에서 상위 5개의 GO term을 분석에 활용하였다.
NAC 유전자의 아미노산 서열 구성 분석
NAC 유전자의 도메인 구조를 분석하기 위해 InterProScan 5(-f tsv-appl Pfam) (Jones et al. 2014)로 생성된 TSV 파일을 활용하였다. 분석의 정확도를 높이기 위해 e-value가 1e-4 이하인 결과만 분석에 포함하였다. NAC 도메인의 서열 특성을 확인하기 위해 MAFFT v.7.470(--maxiterate1000) (Katoh and Standley 2013)로 alignment를 수행하고 TrimAL v1.4(-gt 0.3) (Capella-Gutierrez et al. 2009)를 통해 부정확한 alignment 영역을 제거하였다. EMBOSS cons를 이용해 보존 서열을 확인하고 NAC 도메인의 signature 서열을 확인하였다. 정렬된 서열을 기반으로 WebLogo v2.8.22(Crooks et al. 2004)를 이용하여 아미노산 보존도를 시각화하였으며, Jalview v2.11.2.6(Procter et al. 2021)을 통해 종 간 서열 보존도를 비교하였다. 또한 선행연구(Liu et al. 2023a)를 참고하여 NAC 도메인의 보존 부위와 2차 구조를 예측하였다.
NAC 유전자의 계통학적 분석
NAC 유전자 간의 계통학적 관계를 분석하기 위해 이전 연구에서 사용된 방법을 기반으로 계통수 분석을 수행하였다(Jang et al. 2024). MAFFT v7.470(--maxiterate 1000) (Katoh and Standley 2013)을 사용하여 서열 정렬을 수행한 뒤, TrimAL v1.4(-gt 0.3) (Capella-Gutierrez et al. 2009)로 부정확한 정렬 영역을 제거하였다. IQ-TREE v2.0.6(-alrt 1000 -B 1000) (Minh et al. 2020)을 이용하여 계통수를 생성하고, iTOL v6(https://itol.embl.de/)을 활용하여 시각화하였다. 도메인 및 모티프 분석 결과를 바탕으로 NAC 유전자를 9개의 서브그룹으로 분류하였다.
NAC 유전자의 복제 분석
NAC 유전자의 서브그룹별 복제 및 확장 양상을 분석하기 위해 CAFE v5(De Bie et al. 2006)를 사용하였다. TimeTree(http://www.timetree.org/)에서 확보한 4종의 종간 계통수를 기반으로, 각 서브그룹에 속하는 NAC 유전자 수를 집계하여 CAFE 분석을 수행하였다. 기본 매개변수(p = 0.05)를 사용하였으며, 분석 결과 중 “Base_change.tab” 파일에서 양의 값을 가지는 branch point만을 시각화에 활용하였다.
저온 스트레스 처리 시 배의 NAC 유전자 발현 분석
배 NAC 유전자의 저온 반응성을 확인하기 위해, 기존 연구(Li et al. 2021)에서 보고된 배 꽃눈 RNA-seq 데이터를 5°C 저온 스트레스 조건 하에서 네 시점(0일, 5일, 30일, 45일)에 수집하여 활용하였다. 이전 연구에서 사용된 방법을 기반으로(Kim et al. 2024), CLC Assembly Cell(CLC Bio, Aarhus, Denmark)을 이용하여 FASTQ 파일을 트리밍하고, HISAT2(-dta -x) (Kim et al. 2019)를 사용하여 P. pyrifolia 참조 유전체에 매핑하였다. 이후 StringTie(-e -B -G) (Pertea et al. 2015)를 이용해 FPKM 값을 계산하고, Python 스크립트(prepDE.py3)를 통해 read count로 변환하였다. 차등 발현 유전자(Differentially expressed genes; DEGs)는 DESeq2(Love et al. 2014)를 이용해 |log2FoldChange| > 2 및 adjusted p-value < 0.05 기준으로 선별하였다. 선별된 NAC 유전자 및 DEGs에 대해 R 패키지 Mfuzz(Kumar and Futschik 2007)를 활용하여 클러스터링을 수행하였다. GO term enrichment 분석은 OmicsBox v3.4를 사용하여 진행하였고, BLASTP(nr v5, e-value < 1e-3) 및 InterProScan 결과를 통합하여 기능 주석을 부여하였다. 분석 결과는 R을 사용해 시각화하였다. 공동 발현 네트워크는 R 패키지 WGCNA(Langfelder and Horvath 2008)를 이용해 SFT.R.sq > 0.8 및 minModuleSize = 30 조건의 최적 β값을 선정하여 구축하였으며, 최종 네트워크는 Cytoscape v3.10.3(Shannon et al. 2003)을 통해 시각화하였다.
결과 및 고찰
NAC유전자 정보 업데이트
선행연구에서 동정된 벼, 애기장대, 사과, 배의 NAC 유전자 정보를 업데이트하기 위해 재동정을 수행한 결과 총 677개의 NAC 유전자가 확인되었으며, 벼 152개, 애기장대 112개, 사과 202개, 배 211개로 구성되었다(Table 1 and Supplementary Table 2). 이 중 92개(13.6%)는 새롭게 동정된 NAC 유전자로, 특히 사과 46개(50.0%)와 배 36개(39.1%)를 포함해 신규 NAC 유전자의 89.1%가 장미과 작물에서 확인되었다(Fig. 1A). NAC 유전자의 서브도메인 구조 분석 결과, A-B-C-D-E의 다섯 개 서브도메인을 모두 포함하는 구조가 369개(54.5%)로 가장 많았으며, 그 외 A-B-C-D(47개, 6.9%), A(35개, 5.2%), A-C-D-E(26개, 3.8%), A-C-D(26개, 3.8%) 순으로 존재하였다(Fig. 1B). 이는 NAC 도메인이 보존도가 높은 서브도메인과 보존도가 낮은 C말단 transcriptional activation region(TAR) 영역으로 구성된다는 기존 보고와 일치하였다. 업데이트된 4종의 NAC 유전자의 기능을 추정하기 위해 GO term 분석을 수행한 결과, biological process에서는 “regulation of DNA-templated transcription”, cellular component에서는 “nucleus”, molecular function에서는 “DNA-binding transcription factor activity”가 가장 빈번하게 나타났다(Fig. 1C). 이는 NAC 유전자가 전사 인자로서 전사 조절 및 DNA 결합 기능을 수행한다는 기존 보고와 부합하며, 본 연구에서 재동정된 NAC 유전자들 또한 전사 인자의 역할을 수행할 가능성이 높음을 시사한다.
Table 1.
Numbers of re-annotated NAC genes in four species
| Species | Previously annotated gens | Newly annotated genes | Total |
| Oryza sativa | 142 | 10 | 152 |
| Arabidopsis thaliana | 112 | 0 | 112 |
| Malus domestica | 156 | 46 | 202 |
| Pyrus pyrifolia | 175 | 36 | 211 |
| Total | 585 | 92 | 677 |
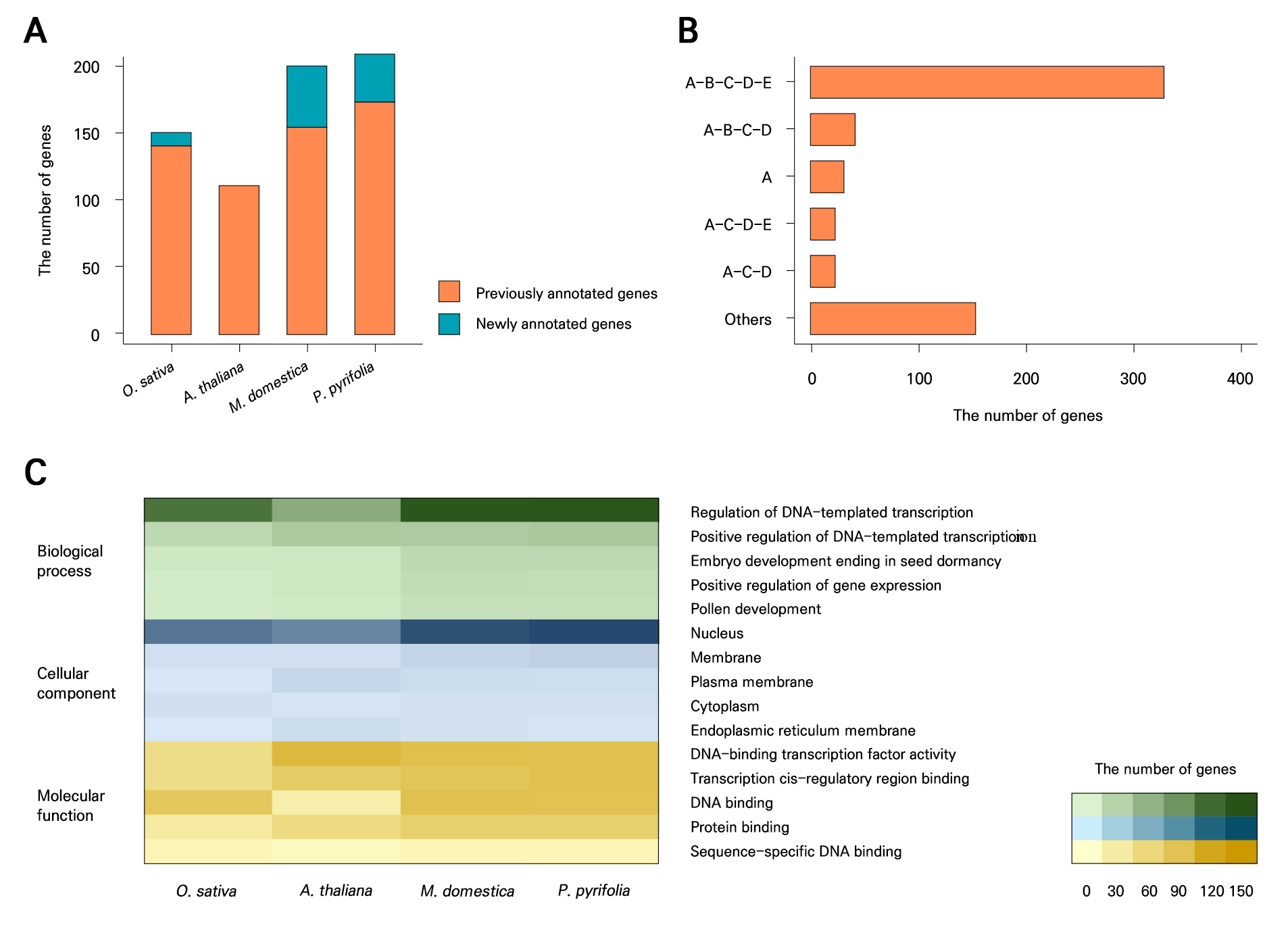
Fig. 1.
Characteristics of NAC genes in four species, Oryza sativa, Arabidopsis thaliana, Malus domestica, and Pyrus pyrifolia: (A) Number of re-annotated NAC genes. Orange and green bars represent previously annotated and newly annotated genes, respectively. (B) Number of NAC genes in each subdomain repertoire. (C) Number of NAC genes associated with the top five gene ontology (GO) terms. Color gradients represent increasing frequency of GO terms in three categories: biological process (green), cellular component (blue), and molecular function (yellow).
NAC 도메인 및 모티프 구성
NAC 유전자의 보존된 서열을 분석하기 위해 NAC 도메인의 아미노산 보존율을 계산한 결과, 각 서브도메인 위치에서는 50-60%의 보존율을 보였다. 이는 보존율이 약 20%에 불과한 도메인 외부 영역보다 높은 수치로, NAC 서브도메인 A-E가 보존도가 높은 서열로 구성되어 있음을 나타낸다(Fig. 2A). 이들 서브도메인은 NAC 유전자군의 기능 수행에 핵심적인 역할을 할 것으로 예상된다. 서브도메인의 보존도와 구조적 특성을 추가로 확인하기 위해 모티프 분석을 수행한 결과, 반복적으로 나타나는 29개 모티프를 제외한 21개 모티프가 총 15개 영역에 분포하였다(Fig. 2B). NAC 유전자는 NAC 도메인과 C말단의 TAR 영역으로 구성되어 있으며, 각 서브도메인은 특정 영역에 대응되었다: A는 영역 1, B는 영역 3, C는 영역 4-5, D는 영역 6-7, E는 영역 8에 위치하였다. 결과적으로, 서브도메인 A-E는 대부분의 NAC 유전자에서 높은 보존도를 보이며 공통 기능 수행에 관여하는 영역으로 판단된다. 반면, 영역 9-15는 보존도가 낮은 TAR 영역에 해당하며, 이는 유전자 간 구조적 다양성과 기능적 분화를 반영할 가능성이 있다.
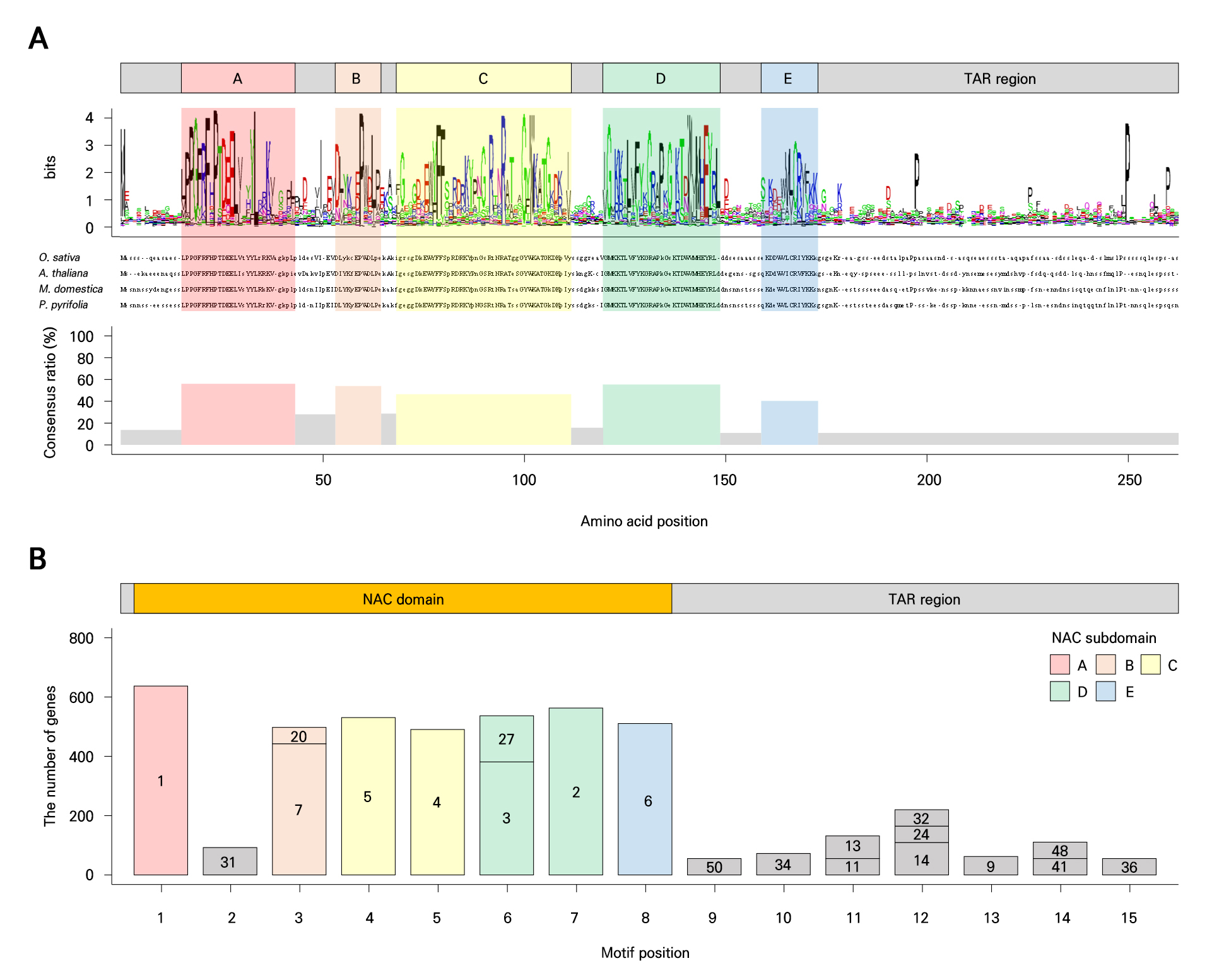
Fig. 2.
Subdomain structure and motif composition of NAC domains from four species: (A) conserved amino acid sequence and five subdomains (A-E) of NAC domains. The x-axis represents the amino acid positions, while the y-axis indicates the relative abundance of each amino acid sequence. The bar graph shows the consensus ratio of eleven compartments corresponding to the five subdomains. (B) Motif composition of NAC genes. Each color represents a specific subdomain, while the remaining motifs are shown in gray.
NAC 유전자들 간의 계통학적 분석
NAC 유전자의 진화적 관계를 분석하기 위해 총 677개의 NAC 유전자에 대해 계통수 분석을 수행하였다(Fig. 3A). 분석에는 O. sativa, A. thaliana, M. domestica, P. pyrifolia 네 종이 포함되었으며, O. sativa와 A. thaliana는 모델 식물로 사용되었다. 유전자 간 모티프 구성, 유연관계, 종 특이성을 기반으로 NAC 유전자를 총 9개의 서브그룹(G1-G9)으로 분류하였다. 각 서브그룹의 유전자 수는 39개에서 131개 사이였으며, 평균 75.2개의 유전자가 포함되었다(Fig. 3B). G4와 G5 그룹에서는 벼의 비율이 각각 34.5%와 43.1%로 장미과 과수에 비해 높게 나타났으며, 반대로 G6에서는 사과(44개, 48.4%)와 배(46개, 50.5%)의 비율이 압도적으로 높고, 벼(0개), 애기장대(1개)는 거의 존재하지 않았다. 이러한 결과는 G6 그룹이 장미과 과수에 특이적으로 존재하는 유전자군임을 시사한다. 서브그룹별 NAC 유전자의 계통 특이적 확장을 분석하기 위해 CAFE 분석을 수행한 결과, G2, G3, G8, G9 등에서 복제 수가 유의하게 증가한 것이 확인되었다(Fig. 3C). 이는 사과와 배가 약 1억 2천만 년 전 공통조상으로부터 분기된 이후, 모델 식물과 구분되는 장미과 과수 특이적인 진화 양상을 보였음을 시사한다. 특히 G8에서 가장 뚜렷한 유전자 확장이 관찰되어, 본 연구에서는 이 그룹을 중심으로 기능적 분석을 수행하였다.
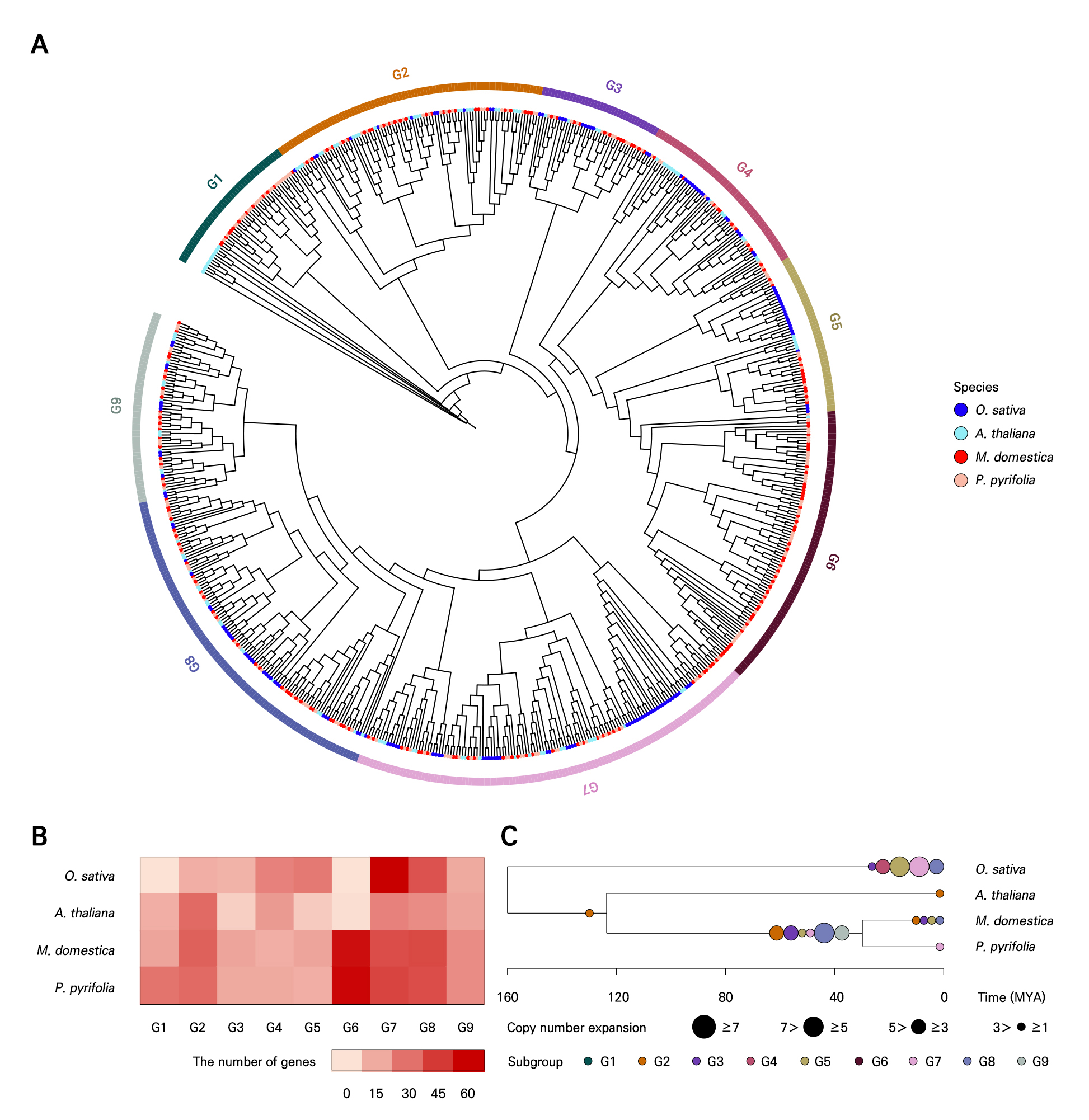
Fig. 3.
Phylogenetic relationships and gene expansion of NAC genes: (A) phylogenetic tree of NAC genes in four species. The dot colors represent the four species. The outer ring indicates nine subgroups. (B) Number of NAC genes in each subgroup. The scale bar at the bottom indicates the number of NAC genes. (C) Copy number expansion of the NAC gene subgroups in each species. The circle colors correspond to the subgroup colors in the phylogenetic tree, and the circle size reflects the degree of gene copy number expansion. Divergence time is shown in MYA (million years ago).
저온 처리 시 유전자 발현 분석
NAC 유전자는 비생물학적 스트레스에 대한 저항성과 관련된 주요 전사인자로, 특히 저온 스트레스 조건에서의 기능이 주목되고 있다. 이를 규명하기 위해 P. pyrifolia를 대상으로 5°C 저온 처리 후 0일, 15일, 30일, 45일의 4개 시점에서 RNA-seq 데이터를 분석하였다. DEGs와 NAC 유전자의 발현 패턴을 비교하여 유사한 발현 양상을 보이는 유전자들을 중심으로 클러스터링을 수행하였고, 총 4개의 클러스터(C1-C4)가 도출되었다(Fig. 4A). 각 클러스터에는 C1 502개, C2 424개, C3 408개, C4 555개의 유전자가 포함되었다. NAC 유전자의 클러스터 분포를 분석한 결과, 저온 반응 클러스터(C2-4)에 장미과 특이적으로 확장된 G6과 G8 서브그룹 NAC 유전자가 다수 포함되어 있었다(Fig. 4B). 구체적으로, C2에서는 G8 유전자 7개, C3에서는 G6 유전자 7개, C4에서는 G8 유전자 12개로 가장 높은 비율을 차지하였다(Fig. 4B). 이러한 결과는 장미과 특이적으로 확장된 G6과 G8 서브그룹이 저온 반응 클러스터와 밀접하게 연결되어 있음을 보여주며, NAC 유전자군의 계통 특이적 확장이 장미과 과수의 저온 스트레스 적응에 기여했을 가능성을 시사한다.
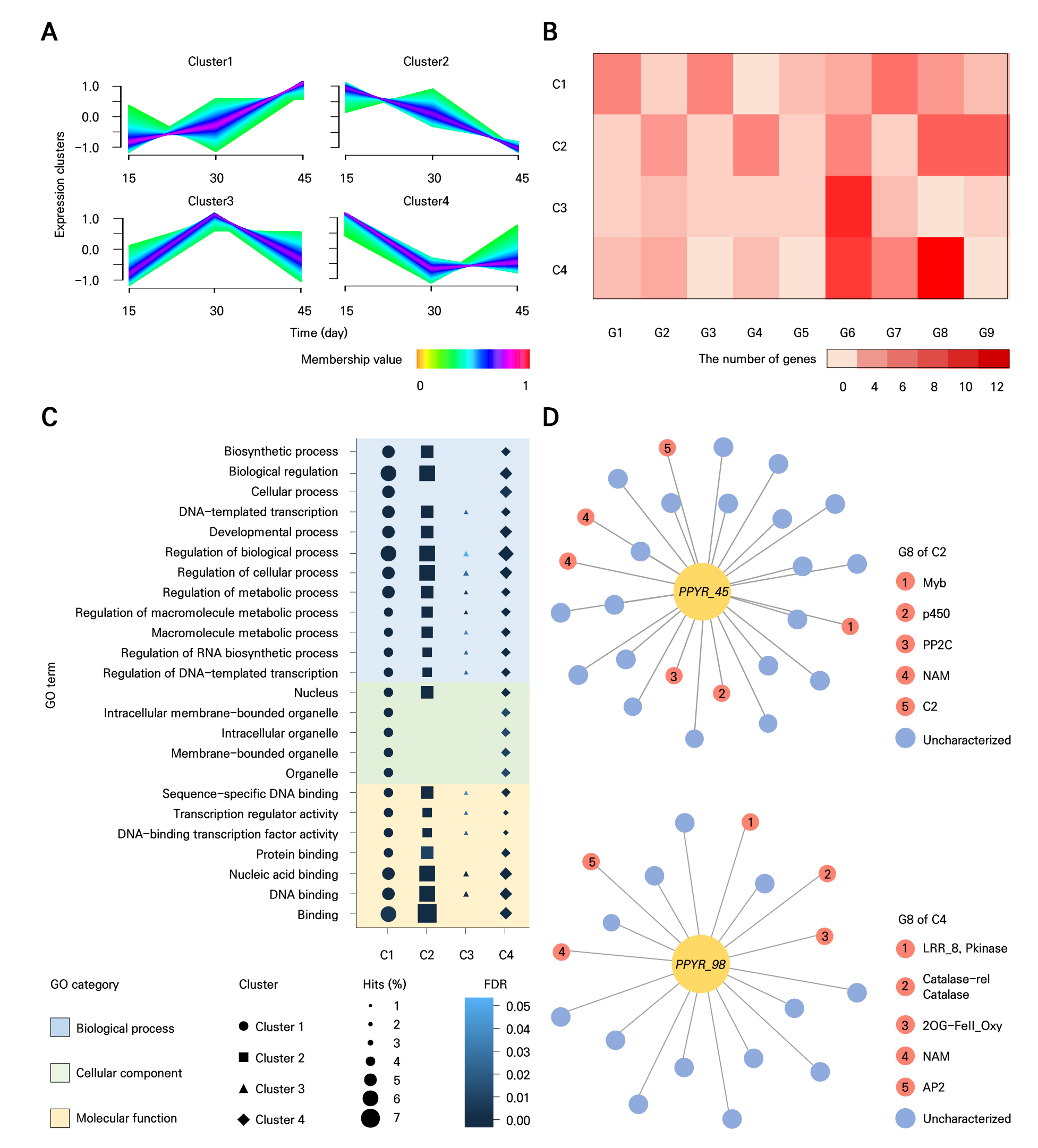
Fig. 4.
Expression analysis of NAC genes in P. pyrifolia under cold stress: (A) expression patterns of differentially expressed genes (DEGs) and NAC genes in response to cold stress. (B) Heat map showing the number of NAC genes from each subset across expression clusters. The scale bar at the bottom indicates the number of NAC genes. (C) Enrichment of GO terms for each expression cluster. The background color represents the three GO categories. Symbol shape and size indicate the frequency of each GO term. (D) Co-expression network of NAC genes under cold stress. Orange nodes represent genes with annotated domains, while blue nodes indicate uncharacterized genes.
GO enrichment 분석 결과, C1, C2, C4에서 “biosynthetic process(GO:0009058)”가 공통적으로 나타났으며, C1-C4 전 클러스터에서 “regulation of cellular process(GO:0050794)”, “regulation of macromolecule metabolic process(GO:0060255)”, “macromolecule metabolic process(GO:0043170)” 등 다양한 저온 스트레스 관련 기능이 확인되었다(Fig. 4C). 특히 “carbohydrate derivative biosynthetic process(GO:1901137)”, “regulation of lipid storage(GO:0010883)”, “regulation of polysaccharide metabolic process(GO:0032881)”, “polysaccharide catabolic process(GO:0000272)”등은 저온 저항성과 밀접한 관련이 있는 하위 GO term으로, 기존 연구에서도 탄수화물 유도체, 지질 저장, 다당류 대사 등이 식물의 저온 적응에 기여하는 것으로 보고된 바 있다(Thomashow 1999; Yano et al. 2005; Bogdanovic et al. 2008; Moellering et al. 2010; Valitova et al. 2019). 이는 세포막 안정성을 유지하는 지질 축적과, 에너지 공급 및 삼투 조절에 관여하는 다당류 대사가 저온 조건에서 생리적 적응에 중요한 역할을 함을 보여주며, 해당 클러스터가 저온 스트레스 대응과 직접적으로 연계되어 있음을 시사한다. WGCNA 분석 결과, NAC 유전자 PPYR_45(Ppy08g0997.1)와 PPYR_98(Ppy10g0753.1)이 공동 발현 네트워크에서 중심 허브 유전자로 기능하고 있었다(Fig. 4D and Supplementary Table 3). PPYR_45와 함께 발현된 유전자 중, Malus baccata의 MbMYBC1(Myb)은 저온 및 가뭄 스트레스 저항성을 높였고(Liu et al. 2023b), Glycine max의 GmZF1(C2)은 저온 유도 유전자인 COR6.6의 발현을 유도하였다(Yu et al. 2014). 또한 Solanum lycopersicum의 SlNAM1과 Capsicum annuum의 CaNAC064(NAM)는 저온 내성 증가에 기여하였으며, 특히 CaNAC064는 knockout 시 감수성이 증가해 기능이 입증되었다(Li et al. 2016a; Hou et al. 2020). Zea mays의 ZmPP2C2(PP2C)도 발아 단계에서 저온 내성을 향상시켰다(Hu et al. 2010). PPYR_98과 공동 발현된 유전자들도 저온 반응과 관련된 도메인을 포함하고 있었다. S. lycopersicum의 SlF3HL(2OG-FeII_Oxy), Triticum aestivum의 CaT(Catalase), C. annuum의 CaPF1(AP2), Z. mays의 ZmMKK1(Pkinase)은 모두 ROS 억제, 막 안정성 강화, 발현 조절을 통해 저온 스트레스 저항성을 부여하는 것으로 보고되었다(Matsumura et al. 2002; Yi et al. 2004; Cai et al. 2014; Meng et al. 2015). 따라서 PPYR_45와 PPYR_98은 장미과 과수의 저온 스트레스 적응을 매개하는 핵심 후보로 간주될 수 있으며, 향후 기능 검증과 분자적 메커니즘 규명을 통해 저온 내성 향상에 활용 가능한 유전자 자원으로 발전하여 품종 개발을 위한 분자육종에 기여할 수 있을 것이다.
종합적으로, 본 연구에서는 벼, 애기장대, 사과, 배의 NAC 유전자를 재동정하고, 장미과 과수 작물 중심으로 구조 및 진화 분석을 수행하였다. 재동정을 통해 총 677개의 NAC 유전자를 확보하였으며, 모티프 분석 결과 N말단 서브도메인은 높은 보존도를, C말단 TAR 영역은 낮은 보존도를 보였다. 이는 NAC 유전자의 기능적 다양성과 진화적 특성을 반영하는 것으로 해석된다. CAFE 분석에서 G8 그룹의 유전자 확장이 확인되었고, GO 분석과 공동 발현 네트워크 결과는 G8에 속한 NAC 유전자들이 저온 저항성과 관련된 기능을 수행할 가능성을 강하게 시사하였다. 특히 PPYR_45, PPYR_98은 관련 유전자들과의 높은 공동 발현을 보이며, 저온 저항성 향상에 기여할 수 있는 핵심 후보 유전자로 제안된다. 본 연구는 장미과 과수의 저온 스트레스 대응 메커니즘 이해에 기여할 뿐 아니라, 향후 분자 육종 연구의 기초 자료로 활용될 수 있을 것으로 기대된다.
Supplementary Material
Supplementary materials are available at Horticultural Science and Technology website (https://www.hst-j.org).
- HORT_20250065_Table_S1.xlsx
Supplementary Table 1. Genomic resources used in this study
- HORT_20250065_Table_S2.xlsx
Supplementary Table 2. List of re-annotated NAC genes
- HORT_20250065_Table_S3.xlsx
Supplementary Table 3. List of cold-responsive co-expression network
