

Introduction
Materials and Methods
Plant material
Genetic analysis
Colorimetry measurements of seed coat colors
DNA isolation
BSA-seq analysis
SNP markers for an HRM analysis
Results
Phenotypic segregation and inheritance of seed coat color
Whole-genome resequencing and mapping
Scanning candidate genomic regions for seed coat color
Localization of the WSC and RSC candidate genomic regions according to SNP markers and physical mapping
Discussion
Introduction
Watermelon (Citrullus lanatus; 2n = 2x = 22) is a globally important vegetable crop. In 2022, South Korea reported a production volume of 476,897.99 tons and a cultivated area of approximately 11,346 ha (FAOSTAT, 2022; https://www.fao.org/faostat/). In 2023, watermelon production in South Korea amounted to 1,569.9 billion South Korean won (KRW), accounting for 4% of the agricultural production total. Additionally, seed production reached 593 kg in total (Korean Seed Association; http://kosaseed.or.kr/).
Watermelon flesh is high in water and rich in phytochemicals such as lycopene, β-carotene, and ascorbic acid. The lycopene content in watermelons is more than 60% higher than that in tomatoes (Guner and Wehner, 2004). Watermelon contains citrulline, a non-essential amino acid that serves as a source of antioxidants and vasodilators. Citrulline is a by-product generated during the oxidation of arginine and plays a role in protecting blood vessels by delivering nutrients to cells in the cardiovascular system (Rimando and Perkins-Veazie, 2005). Watermelon seeds are also rich in proteins that serve as precursors of antioxidant peptides (Wen et al., 2020). According to previous studies, watermelon seed protein hydrolysates with molecular weights less than 1 kDa exhibit strong antioxidant capabilities (Wen et al., 2019a).
Seed color affects various physiological processes, such as water uptake (Powell, 1989; Mavi, 2010), gas diffusion (West and Harris, 1963), seed dormancy (Baskin et al., 2000), and overall seed quality, owing to certain pigments in the seed coat (Powell, 1989; Mavi, 2010). Seed coat color is linked to the biochemical properties of seeds, including the antioxidant levels and activities (El-Bramawy et al., 2008), and affects the appearance of the flesh as well (Wehner, 2008). Black seeds are attractive when paired with red or canary yellow flesh, whereas near-seedless cultivars show white or light-colored seeds (Wehner, 2008). Among these traits, the coat color of watermelon seeds is of significant economic value, particularly in Asia and West Africa. In these areas, watermelon seeds are cultivated not only for their horticultural qualities but also for their culinary uses in snacks and soups, making seed coat color a critical factor in relation to consumer preferences and marketability (Gusmini et al., 2004; Mavi, 2010).
Watermelon seed coat colors exhibit a wide range of variations, including solid black, dotted black, brown, tan, green, red, white, and black stripes with white (Poole, 1941; Poole, 1944; Paudel et al., 2019). Poole et al. (1941) investigated a genetic model for controlling seed coat color by examining 40 different segregated populations. Their study suggested a three-gene model involving genes R, T, and W, along with a modifier gene, D.
With the advent of next-generation sequencing (NGS) technology, sequencing has become a high-throughput, less error-prone and more cost-effective method. Consequently, NGS has been used extensively to identify molecular markers across genomes (Ruangrak et al., 2018). The publicly available watermelon reference genome includes the watermelon cultivar 97103 and the American heirloom variety Charleston Gray. These genomes are available in the Cucurbit Genomics Database, which provides essential resources for molecular marker development, gene mapping, quantitative trait locus (QTL) mapping, and molecular breeding research (Guo et al., 2013; Guo et al., 2019; Wu et al., 2019). A relatively recent advancement in exploring major QTLs is the bulked segregant analysis combining NGS data (BSA-seq; also known as QTL-seq) to identify the QTLs and genetic markers needed for marker-assisted selection (MAS) (Michelmore et al., 1991; Takagi et al., 2013). A major benefit of BSA-seq is that it does not require the genotyping of every individual within a population, thus significantly simplifying the process. In watermelons, BSA-seq was initially used to map a dwarfism locus on chromosome (Chr.) 7 (Dong et al., 2018). This technique has been used to rapidly develop molecular markers of various watermelon traits to enhance breeding efficiency. Additionally, BSA-seq has been used to identify genetic loci in watermelons and other crops, highlighting its versatility and effectiveness (Takagi et al., 2013; Jang et al., 2019; Wen et al., 2019b; Jang et al., 2020; Sugihara et al., 2020; Vogel et al., 2021; Wang et al., 2021). Using BSA-seq, researchers can rapidly identify crucial genetic markers, facilitate targeted breeding efforts, and improve crop traits.
Based on NGS-based BSA-seq and genotyping with sequencing (GBS), Paudel et al. (2019) used three segregated F2 populations (dotted black × green, dotted black × red, and dotted black × clump) to map the R, T1, W, and D loci on chromosomes 3, 5, 6, and 8, respectively. They developed allele-specific PCR (KASP™) assays and single-nucleotide polymorphism (SNP) markers for genotyping (Paudel et al., 2019). Additionally, Li et al. (2020) utilized an NGS-based genetic mapping approach with a light yellow and black seed coat RIL population to identify Cla019481 on chromosome 3 as the gene responsible for black seed coat color (Li et al., 2020). In 2022, Maragal et al. employed a BC1F2 mapping population of BIL-53 (Citrullus amarus, red) × IIHR-140-152 (C. lanatus, black) to identify three QTLs related to seed coat color: q_SCC_3.1, q_SCC_5.1, and q_SCC_5.2. Although several studies have been conducted, a genetic analysis using mapping populations generated by crossing parental lines harboring red and white seed coat colors in watermelons remains unexplored.
In this study, we aimed to identify candidate genomic regions in the red and white seed coats of watermelon using BSA-seq. The resulting delta SNP indices were analyzed by comparing sequences between two bulks from plants harboring red and white seed coats, and a SNP-based marker linked to seed coat color was developed by means of a high-resolution melting (HRM) analysis. This research provides insights into the genomic regions determining seed coat color in watermelon, facilitating marker-assisted seedling selection in watermelon breeding programs.
Materials and Methods
Plant material
The female and male parental lines used in this study were RG-21 (C. lanatus) (Fig. 1A) and Wr-609 (C. amarus) (Fig. 1B), which had white and red seed coats, respectively. Plant cultivation and seed production were conducted in a greenhouse using Partner Seeds (Gimje, South Korea). An F1 hybrid was produced by crossing them in the summer of 2018 to create a mapped population. Thereafter, an F2 generation was created by self-pollinating F1 individuals in the summer of 2019. From the F2 population, 219 individuals were grown for phenotypic surveys (Fig. 1C), and leaf samples were collected for DNA extraction.
Genetic analysis
We selected clear seed coat color phenotypes (n = 80) from the F2 progeny for an inheritance analysis using a colorimetric measurement method. The goodness-of-fit between the expected and observed segregate ratios was analyzed using the chi-square test in R software (R Core Team, 2021).
Colorimetry measurements of seed coat colors
Intact seed coat colors were measured using a 3nh NR310 colorimeter (Shenzhen 3NH Tech. Co., Guangzhou, China). The light source was D65, and the excitation device was an LED blue light. Each measurement consisted of CIE L*, a*, and b*, representing lightness and red/green and blue/yellow colors, respectively. Each seed was measured ten times, and the mean value was used in the data analysis. Based on these measurements, C* (chroma: saturation of the color) and hab (hue angle: purity of the color) were calculated using the following equations:
Principal component analysis (PCA) was utilized to assess the values of CIE L*, C*, and hab with the FactoMineR package (Lê et al., 2008), with visualization conducted using the ggplot2 package (Wickham, 2016) in R software ver. 4.1.2 (R Core Team, 2021).
DNA isolation
Leaf samples were collected for DNA isolation from individually tagged plants grown in a greenhouse (before they matured sufficiently). Genomic DNA was extracted using the GeneAll® Exgene™ Plant SV mini kit (GeneAll® Biotechnology, Seoul, Korea) in accordance with the manufacturer’s instructions, with appropriate modifications. The quality of the extracted DNA was monitored using a Nanodrop spectrophotometer (DS-11, Denovix, Wilmington, DE, USA) and 1.2% agarose gel electrophoresis. The purified DNA samples were quantitatively analyzed using a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA) using Qubit™ dsDNA HS assay kits (Invitrogen), diluted to a working dilution of 20 ng/µL, and stored in a refrigerator at −20°C.
BSA-seq analysis
In this procedure, 30 plants with an extremely white seed coat (WSC) phenotype and 30 plants with an extremely red seed coat (RSC) phenotype were selected from the F2 progeny for BSA, and 5 µg DNA was pooled from each individual to create WSC and RSC bulks. The two bulk and parental lines were sequenced at a sequencing depth of approximately 30× using a NovaSeq 6000 system (Illumina, San Diego, CA, USA). The resulting ‘fastq’ files were filtered using FastQC (Wingett and Andrews, 2018), and adapter sequences were eliminated using Trimmomatic v 0.39 (Bolger et al., 2014). High-quality paired reads obtained were aligned to the watermelon (97103) v2 genome (Guo et al., 2019) by means of Burrows–Wheeler alignment (BWA ver.0.7.17-r1188) with the mem option (Li and Durbin, 2009). Inaccurate paired reads were filtered, sorted, and deduplicated using “samtools” (Li et al., 2009) to produce BAM files. The variant call format (VCF) was generated via the “mpileup” command using BCF tools (Li, 2011). The VCF was filtered based on a sequencing depth of >30, a minor allele frequency of >0.01, and a missing sequence rate of 10%. The SNP index was estimated as the genomic difference between the WSC and RSC bulk and filtered by homo-SNPs in the WSC parental line of RG-21. Plots with options, including a 0.2 Mb window size and a 10 kb step size, were created using the QTL-seq program (Takagi et al., 2013). Positive and high SNP indices are significant genomic areas with seed coat color characteristics (Sugihara et al., 2020). Parental line sequences were used to identify hetero-SNPs and to construct PCR primers for an HRM investigation (Jang et al., 2020).
SNP markers for an HRM analysis
A genomic area with a high SNP index was determined to have a substantial QTL, and the Primer3 web tool (https://bioinfo.ut.ee/primer3-0.4.0/) was used to design the SNP-based primer sets (Rozen and Skaletsky, 2000). For the HRM analysis, PCR was performed using the LightCycler 96 platform (Roche, Mannheim, Germany). In a volume of 10 µL, 2 ng of dsDNA, 1× LightCycler 480 High-Resolution Melting Master (Roche), 0.5 µM of each primer, and 3 mM MgCl2 were included in a reaction under the following conditions: 55 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The default HRM option was used for the final melting condition. HRM data were analyzed using the LightCycler® 96 SW 1.1 program (Roche Diagnostics, Mannheim, Germany). The amplified products were analyzed via electrophoresis on a 1.5% agarose gel containing 1× TAE (Tris-acetate-EDTA) buffer at 150 V for 30 min, stained with ethidium bromide, and examined under UV light.
A total of 62 F2 progeny previously used for bulk preparation with 34 HRM primer sets were examined to detect significant SNPs located at Chrs. 5 and 6 and to evaluate the significant genomic regions harboring a high ΔSNP index in the BSA-seq analysis. Next, 19 HRM primer sets were prepared and 136 additional F2 progenies were analyzed to test for co-segregation between the tested markers and the seed coat color.
Results
Phenotypic segregation and inheritance of seed coat color
The overall phenotypes of the seed coat color were largely classified into white (with a tan tip), tan, and red in the family (Fig. 1). All seeds from the F1 generation, in which color was absent from the parental lines, had tan seed coats (TSC) only (Fig. 1C and Table 1). The variability in the seed coat color of watermelons due to the interbreeding between RG-21 (C. lanatus with a white seed coat) and Wr-609 (C. amarus with a red seed coat) resulted in inconsistent seed maturity and reduced seed production. This motivated the selection of 80 individuals of F2 progeny required for a meticulous analysis of color segregation, ensuring the representation of seed coat colors of the maternal, paternal, and F1 generations. The seed coat colors were segregated into 14 white, 48 tan, and 18 red individuals. Based on the digitalized optical phenotyping results obtained from the F2 progeny, we conducted a two-dimensional PCA to determine the variance in the segregated individuals (91.22%) explained by each principal component: PC1 (63.05%) and PC2 (28.17%) (Fig. 2). In the PCA visualization, the segregated F2 individuals clustered into three groups and were separated with a difference in the measured CIE L*, C*, and habvariables between the groups. The first group exhibited a red seed coat, which was phenotypically assigned to the color of the parental line Wr-609. The second group had a tan-seed coat corresponding to the F1 phenotype. The third group had a white seed coat that matched that of the parental line RG-21. PCA confirmed that these groups clustered in alignment with the phenotypes observed in the F2 individuals analyzed in this study.

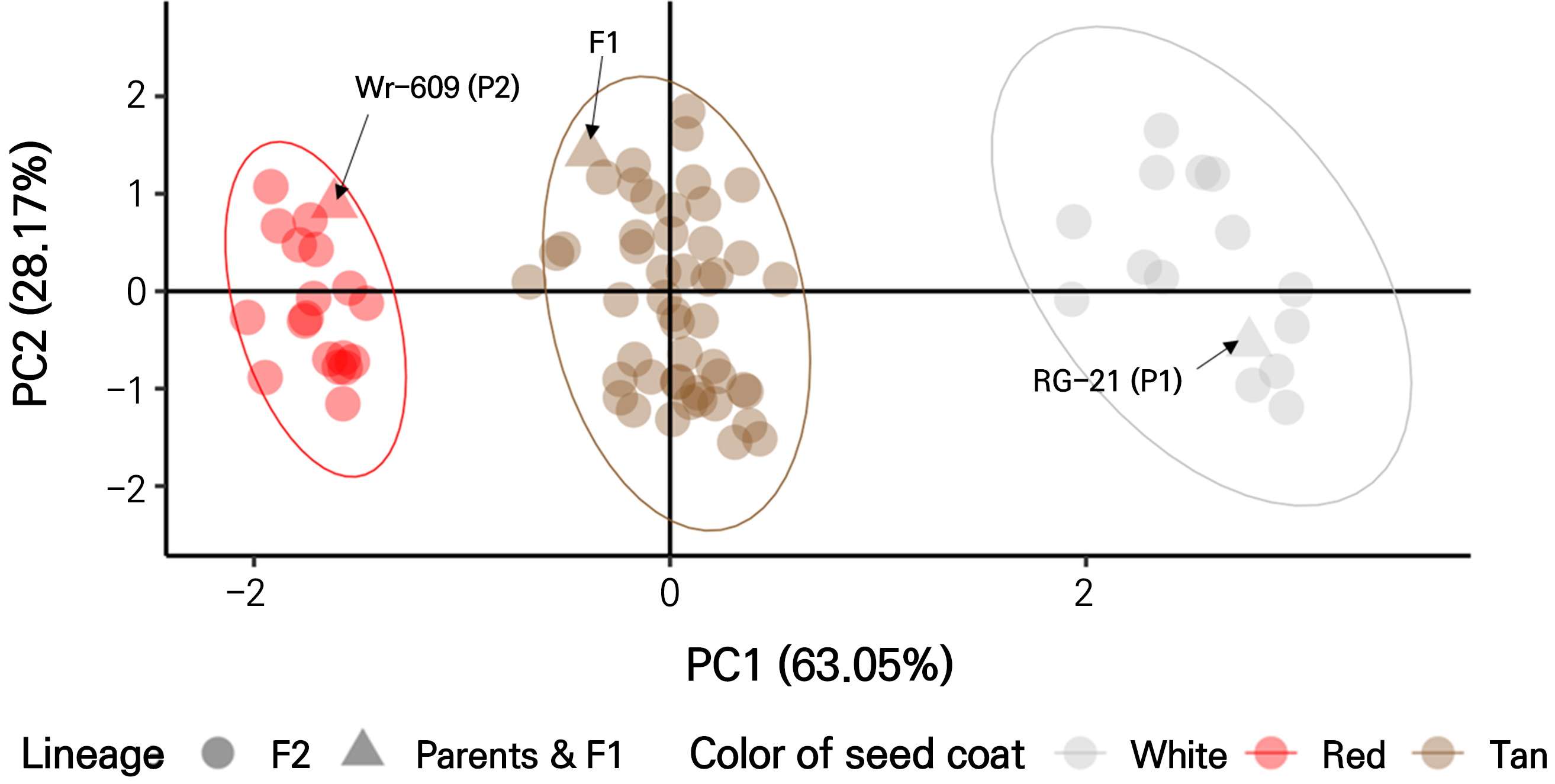
Fig. 2.
Principal component analysis (PCA) analysis clustering of the F2 phenotype based on seed coat color. The three main genotypic groups within the F2 segregating population, as revealed by phenotyping, were as follows: the red seed coat group, phenotypically corresponding to the parental line Wr-609; the tan seed coat group, matching the F1 phenotype; and the white seed coat group, aligned with the parental line RG-21. PCA analyses clustered these groups in accordance with the observed phenotypes in the F2 individuals analyzed in this study.
The segregation ratio for the F2 progeny inferred according to the previously reported inheritance theory (Poole et al., 1941) for seed coat color is typically 9:3:3:1 for tan (RrWw), red (rrWW), white with a tan tip (RRww), and white with a pink tip (rrww). However, in our study, we observed white seed coats without colored tips, leading us to hypothesize a modified segregation ratio of 9:3:4. To test this hypothesis, we conducted a chi-square analysis (α = 0.05) using the results from PCA as the basis for our assumption. The result indicated that the hypothesis of 9:3:4 segregation is not rejected (X2 = 2.6, p-value = 0.2725; Table 1), suggesting that seed coat color is controlled by two genes with negative epistasis.
Table 1.
Phenotyping of seed coat color and chi-square test results of F2 populations (RG-21 × Wr-609, n = 80)
| Generation | Number of seed coat color | Expected | df | X2 | p-valuez | ||
| White | Tan | Red | |||||
| P1 (RG-21) | 30 | 0 | 0 | ||||
| P2 (Wr-609) | 0 | 0 | 30 | ||||
| F1 (RG-21 × Wr-609) | 0 | 30 | 0 | ||||
| F2 (RG-21 × Wr-609) | 14 | 48 | 18 | 4:9:3 | 2 | 2.6 | 0.2725 |
Whole-genome resequencing and mapping
We aimed to understand the genetic inheritance through further research, in this case a BSA-seq analysis. Table 2 displays the whole-genome resequencing and paired-mapping rates. When matched with the reference watermelon genome, the total reads produced for the WSC and RSC bulks exceeded 80 million, and the appropriate paired-mapping rate was greater than 86% (Guo et al., 2019). When comparing BSA bulks at the genome level, the average depth for all covered regions containing high-quality reads was greater than 34, which was considered adequate (Abe et al., 2012; Jang et al., 2020).
Table 2.
Whole-genome re-sequencing and mapping statistics of parental lines and the two bulks of white seed and red plants
Scanning candidate genomic regions for seed coat color
The final 380,726 genomic variations between the WSC and RSC bulks from the F2 progeny were filtered using RG-21 (WSC) sequences as controls to determine the SNP index (Takagi et al., 2013; Itoh et al., 2019). The major loci on Chr. 5 (3.6–7.7 Mb, p < 0.05) and 6 (5.1–9.7 Mb, p < 0.05) were discovered between Chr. 1 and 11 of the watermelon genome when we simulated the average SNP index in a 2.0 Mb interval using a 10 kb increment sliding window (Fig. 3 and Suppl. Fig. S1).
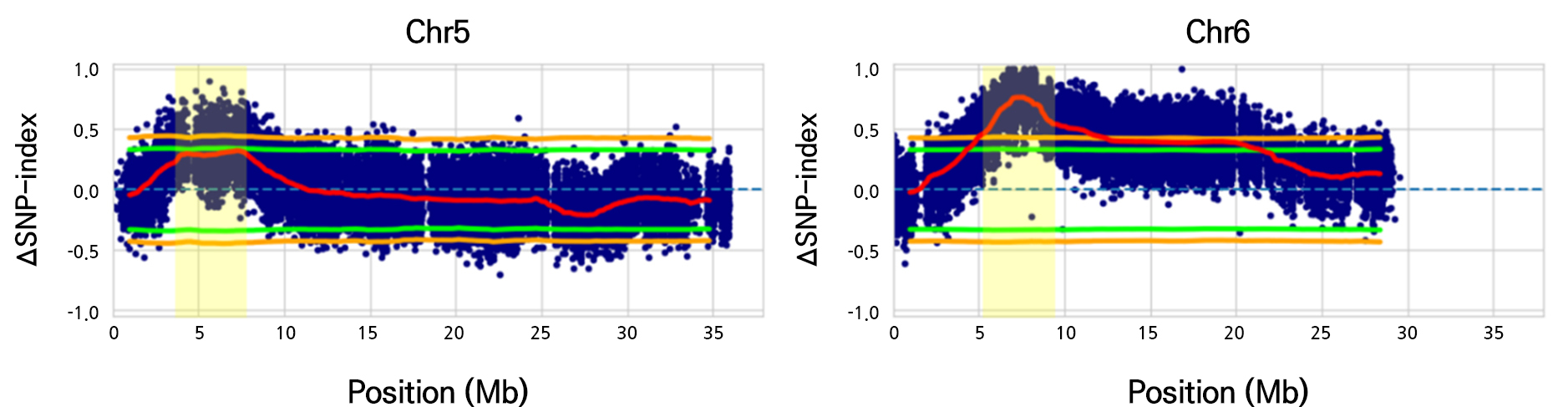
Fig. 3.
Mapping of candidate genomic regions for seed coat color on chromosomes 5 and 6. The ΔSNP index represents the difference between the SNP index of the white seed coat bulk and the red seed coat bulk. Significant candidate genomic regions are enclosed by dotted boxes and are highlighted in yellow. The red line represents the ΔSNP index; the orange line indicates p < 0.01; the green line denotes p < 0.05.
Localization of the WSC and RSC candidate genomic regions according to SNP markers and physical mapping
In the initial BSA-seq analysis, we designed 34 HRM primer sets targeting regions with distinct variants from the parental line, ranging from 3.6 Mb to 7.7 Mb on Chr. 5 and from 5.1 Mb to 9.7 Mb on Chr. 6. We then tested these primer sets on 60 F2 progeny individuals, which were used for bulk preparation in the BSA-seq analysis, to focus on the candidate regions of Chr. 5 and Chr. 6. To narrow the range of the two candidate loci further, we analyzed nine additional primer sets on Chr. 5 and ten additional primer sets on Chr. 6 in 106 F2 progeny. In the RSC case, the highest co-segregation ratio of SNP markers (92%) ranged from 4,542,806 to 4,700,480 bp on Chr. 5 (Table 3). In the WSC trait, we found that the SNP marker located at 7,008,987 to 7,121,807 on Chr. 6 matched the WSC phenotype perfectly (Table 3).
Table 3.
Validation of the high-resolution melting (HRM) analysis of 19 single-nucleotide polymorphism (SNP) markers
| Chromosome | Location (bp) | SNP | Primerz | Co-segregation ratio (%) |
| 5 | 4,218,433 | C > A | R5-421 | 82 |
| 4,542,806 | G > A | R5-454 | 92 | |
| 4,700,480 | T > C | R5-470 | 92 | |
| 4,852,402 | G > T | R5-485 | 85 | |
| 5,101,217 | A > G | R5-510 | 67 | |
| 5,610,509 | A > G | R5-561 | 58 | |
| 6,446,553 | G > A | R5-644 | 58 | |
| 6 | 6,841,088 | C > T | W6-684 | 89 |
| 6,928,966 | C > T | W6-692 | 95 | |
| 7,008,987 | G > T | W6-700 | 100 | |
| 7,121,807 | C > A | W6-712 | 100 | |
| 7,403,446 | C > T | W6-740 | 92 | |
| 7,566,534 | C > T | W6-756 | 85 | |
| 7,826,437 | T > A | W6-782 | 85 |
zPrimer sequences are provided in Suppl. Table S1.
The physical fine-mapping results identified candidate genomic regions associated with seed coat color in watermelon. In Fig. 4, fine mapping pinpoints a specific interval between 3.6 Mb and 7.7 Mb on Chr. 5, narrowing down to a region of 557,168 bp associated with red seed coat color. Additionally, in Fig. 5, the region was delimited to an interval between 5.1 Mb and 9.7 Mb on Chr. 6, narrowing to a region of 207,137 bp associated with white seed coat color. Genotyping of the F2 progeny and parental lines revealed a clear segregation pattern that aligned with the phenotypic expression of seed coat color. The red seed coat (genotype rr) and white seed coat (genotype ww) phenotypes were predominantly linked to the identified candidate regions, as indicated by the consistent presence of red and white genotype segments in individuals expressing red and white seed coat colors (Figs. 4 and 5).
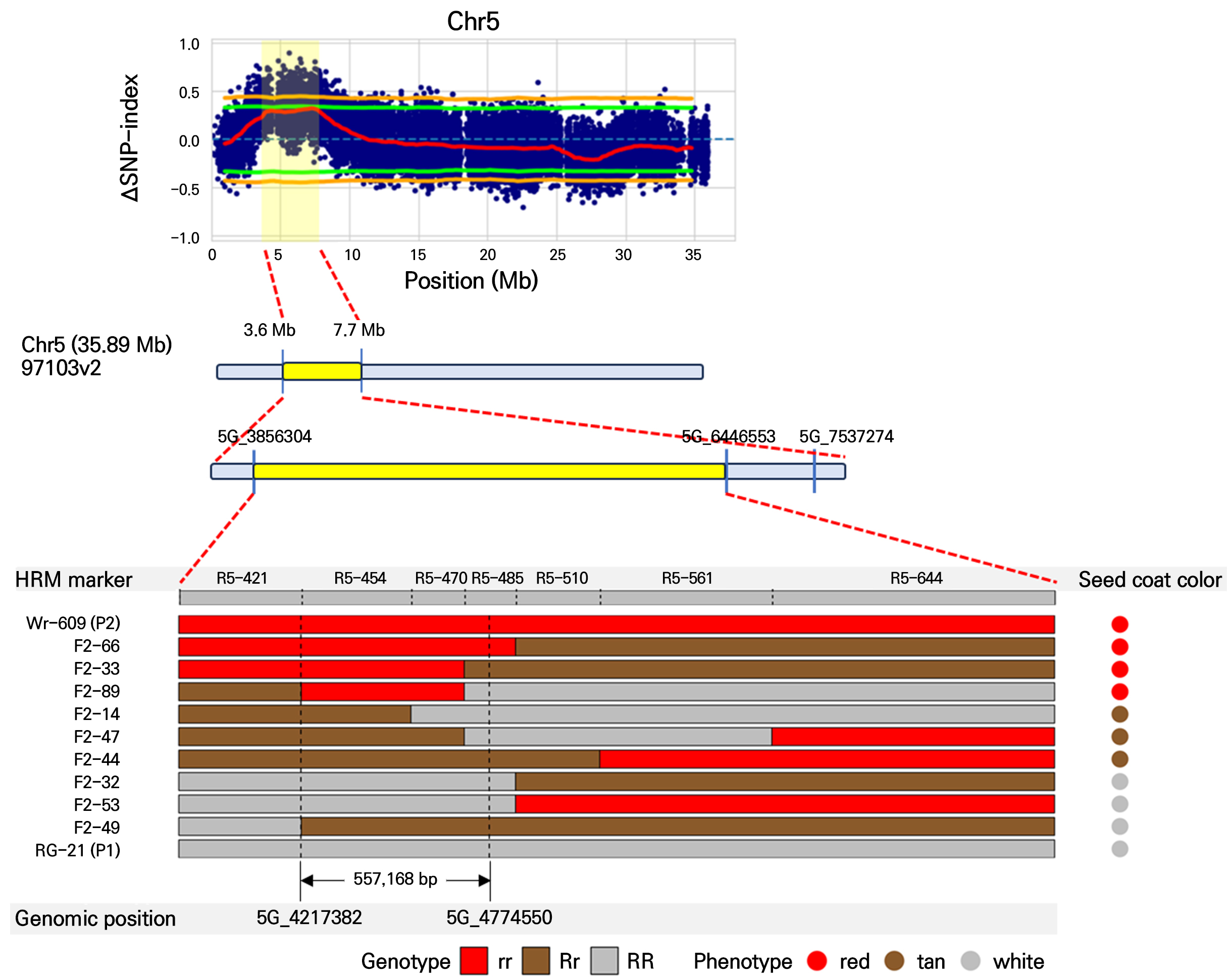
Fig. 4.
Physical fine mapping of the genomic region associated with red seed coat color on chromosome 5 in watermelon. The ΔSNP index plot shows the difference between the SNP index of the white seed coat bulk and the red seed coat bulk along chromosome 5. The significant candidate regions are highlighted in yellow. The physical fine mapping narrows down the candidate region further to a specific interval between 3.6 Mb and 7.7 Mb, with a delimited region of 557,168 bp. Genotyping of the F2 progeny and parental lines revealed a clear segregation pattern corresponding to the phenotypic expression of seed coat color. Red segments indicate the red seed coat phenotype (genotype rr), and tan and white segments correspond to the tan (Rr) and white (RR) phenotypes, respectively.
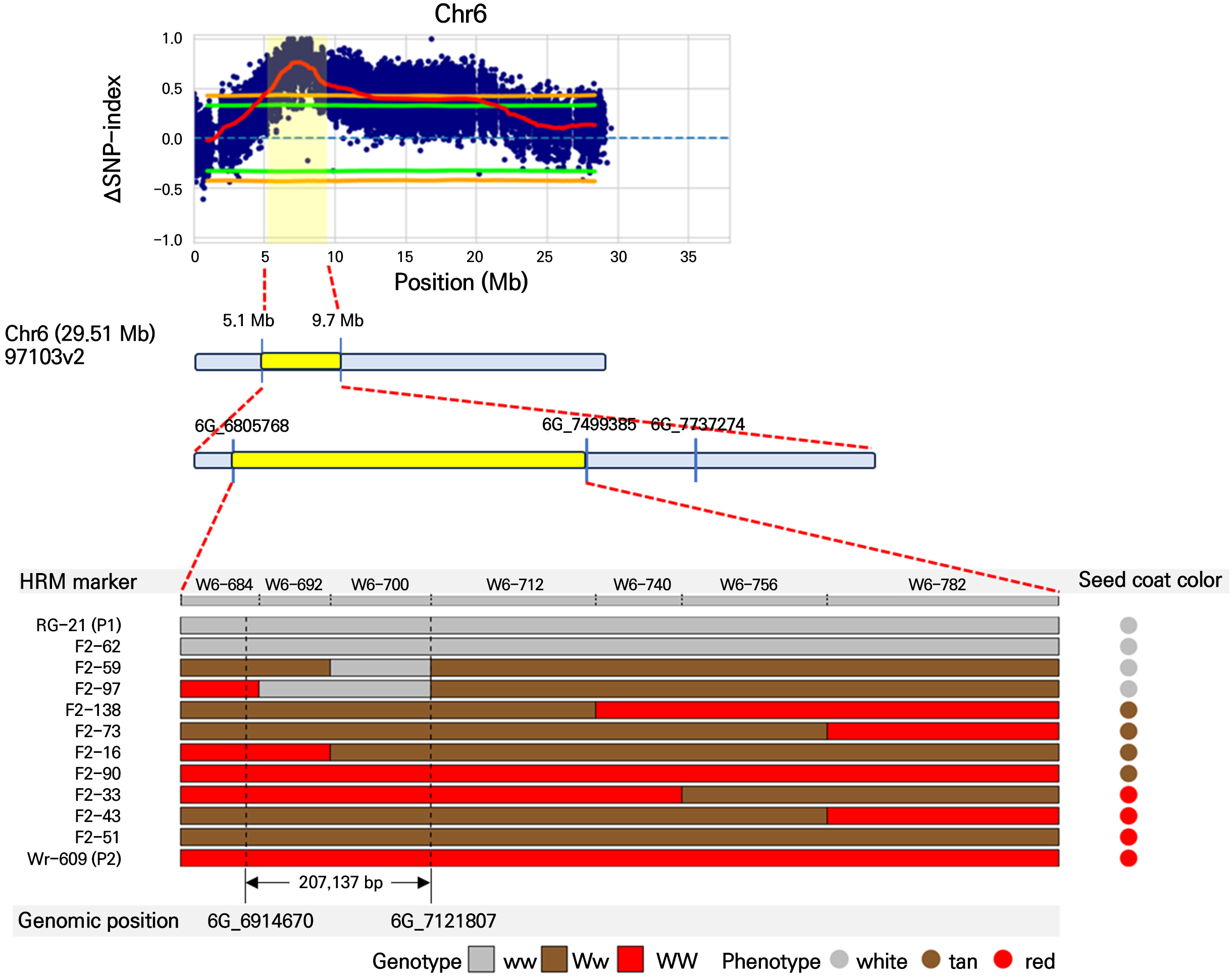
Fig. 5.
Physical fine mapping of the genomic region associated with white seed coat color on chromosome 6 in watermelon. The ΔSNP index plot shows the difference between the SNP index of the white seed coat bulk and the red seed coat bulk along chromosome 6. The significant candidate regions are highlighted in yellow. The physical fine mapping narrows down the candidate region further to a specific interval between 5.1 Mb and 9.7 Mb, with a delimited region of 207,137 bp. Genotyping of the F2 progeny and parental lines revealed a clear segregation pattern corresponding to the phenotypic expression of seed coat color. White segments indicate the white seed coat phenotype (genotype ww), tan segments correspond to the tan phenotype (Ww), and red segments represent the red phenotype (WW).
Discussion
Several genes with intricate genetic connections are responsible for the genetic determination of seed coat color (Poole, 1941). In the first investigation into the inheritance of seed coat color (Kanda, 1931), flat black seed coat color was found to be monogenically dominant over dotted black seed coat color by crossing flat black watermelon seeds with dotted black watermelon seeds. Later, McKay (1936) developed tan red and green red hybrids and demonstrated that the former phenotype is monogenically dominant over the latter in each cross (McKay, 1936). The dotted black seed coat is monogenically dominant over the clump seed coat according to Weetman (1937), who also demonstrated that various combinations of the two genes create clump and tan seed coat colors (Weetman, 1937).
A genetic model consisting of the four genes, R, T, W, and D, was developed in the 1940s to explain the inheritance of seed coat color in watermelons (Poole, 1941). Poole et al. (1941) created 40 distinct segregating populations and suggested a four-gene hypothesis for determining watermelon seed coat color. The seed coat colors produced by various combinations of the R, T, and W genes with the modifier gene D are flat black (RTWD), dotted black (RTWd), green (rTW), tan (RtW), clump (RTw), red (rtW), white-tan tip (Rtw), and white-pink tip (rtw) (Paudel et al., 2019). Additionally, Chrs. 3, 5, 6, and 8 were assigned to the R, T, W, and D genes, respectively. The seed color markers UGA3 5820134, UGA5 4591722, UGA6 7076766, and UGA8 22729513 were also created. In a recent study, the watermelon seed coat was bright yellow or black due to premature termination of protein translation due to a genetic mutation in the 70.2 kb region of Chr. 3 (Li et al., 2020).
In this study, the seed coat color of all F1 hybrids from the cross between RG-21 (WSC) and Wr-609 (RSC) was tan. According to the seed coat color theory for watermelon (Poole et al., 1941; Paudel et al., 2019), the expected genotype for F1 hybrids is RrttWwdd, derived from parental lines RG-21 (RRttwwdd, WSC) and Wr-609 (rrttWWdd, RSC). We did not consider the T and D loci in our analysis, as phenotypes such as flat black (RRTTWWDD), dotted black (RRTTWWdd), or clump (RRTTww) were absent. Consequently, the expected segregation ratio in the F2 generation from a dihybrid cross of F1 (RrWw) should typically follow a 9:3:3:1 pattern for tan, red, white with a tan tip, and white with a pink tip. However, our study found that the phenotypes white with a tan tip (RRww) and white with a pink tip (rrww) were indistinguishable, with only white with a white tip being observed. We hypothesized that negative epistasis between the R and W loci could alter the expected ratio to 9:3:4 (tan, red, white). The BSA-seq analysis suggested that each of these loci (R and W) is located on different chromosomes, lending support to this hypothesis. Upon a closer examination, we found that the genomic regions closely linked to the WSC phenotype were located at 6,914,670 and 7,121,807 bp on Chr. 6. These SNP markers for WSC were closely located in the genomic region controlling the W gene according to a previous report (Paudel et al., 2019). Hence, red in the RSC was closely linked to 4,217,382-4,774,550 bp on Chr. 5, to which the T1 locus for tan was assigned on Chr. 5 (Paudel et al., 2019). Although no variation in the tan phenotype in F1 seeds was observed, the tan phenotype of the F2 progeny varied, i.e., was not fixed. These results imply that the RSC gene located on Chr. 5 could regulate or interact with the T1 locus reported by Paudel et al. (2019). In previous studies, no specific molecular evidence supporting the red color or the genes involved in the red color could be found. Therefore, this study provides a basis for understanding the RSC traits of watermelons and for breeding new watermelon varieties.
Supplementary Material
Supplementary materials are available at Horticultural Science and Technology website (https://www.hst-j.org).
- HORT_20240060_Fig_S1.pdf
Supplemental Fig. S1. Genome-wide result of the bulked segregant analysis with next-generation sequencing to find the causal region for seed coat color. Each panel shows the results of the ΔSNP-index per chromosome. Red line, ΔSNP-index; Orange line, p < 0.01; Green line, p < 0.05
- HORT_20240060_Table_S1.pdf
Supplemental Table S1. Information on genotyping markers for seed coat color in Chrs. 5 and 6
