Introduction
Materials and Methods
Plant Materials, Genome Amplification, and Sequencing
Genome Assembly, Annotation, and Analysis
Phylogenetic Analysis
Results and Discussion
Characteristics of the Leontopodium leiolepis and Anaphalissinica cp Genomes, and Comparison with Other Asteraceae cp Genomes
Introduction
The tribe Gnaphalieae (Cass.) Lecoq & Juillet (i.e., everlasting or paper daisies) comprises 185 genera and 1,240 species and is part of the subfamily Asteroideae in Asteraceae (Ward et al., 2009). In particular, species within the genus Leontopodium are commonly known as ‘edelweiss’. These ornamental plants are grown in gardens and parks (Harrison et al., 2010) and serve as national symbols for various alpine countries, such as Switzerland and Austria. Wild edelweiss plants are protected in several European and Asian countries, and hence they are now being cultivated to conserve their genetic resources (Zheljazkov and Craker, 2016). Therefore, clear taxonomic delimitations are essential for effective breeding and cultivation of these species. The tribe Gnaphalieae, one of the largest and most complex in Asteraceae, was originally regarded as an element of the tribe Inuleae (Merxmüller et al., 1977), but is now considered independent and a sister clade to Anthemideae and Astereae (Panero and Funk, 2008). This reclassification was robustly supported by morphological (Anderberg, 1989; Karis, 1993) and molecular data (Kim and Jansen, 1995; Bayer and Starr, 1998; Wagstaff and Breitwieser, 2002). Despite numerous analyses of the tribe Gnaphalieae, intergeneric relationships are unclear and widely disputed, which is attributed to various factors such as morphological similarity and a complex evolutionary history (e.g., recent rapid radiation and allopolyploidy). Indeed several elements, such as Helichrysum Mill, Pseudognaphalium Kirp., and Philyrophyllum O. Hoffmann & Prantl, have been transferred or split into other genera and/or new tribes (Bentley et al., 2015; Nie et al., 2015).
Phylogenomics is an effective means of addressing complex phylogeny (Bock et al., 2014). Nuclear DNA (nrDNA) has been used extensively to study phylogeny in the tribe Gnaphalieae, whereas chloroplast DNA (cpDNA) analyses based on the characterization of a few loci of have been less useful due to their low resolution, especially in the crown radiation group that comprises most of the tribe Gnaphalieae species. However, recent advances in next-generation sequencing technologies have allowed researchers to obtain numerous complete chloroplast (cp) genome sequences. This rich source of sequence data describing cp genomic structure and gene contents has provided a novel means of studying phylogenetic relationships as well as determining the functional evolution of cp genomes in land plants. Such data has been especially applicable for groups such as Asteraceae that have complex evolutionary histories. For example, the earliest chloroplast-mapping studies revealed two large inversions (22.8 kb and 3.3 kb in the large single copy, or LSC, region) that demonstrated an ancient evolutionary split within Asteraceae (Kim et al., 2005). Several recent comparative analyses have also detailed some structural changes in Asteraceae (e.g., inversion of the small single copy, or SSC, region; Liu et al., 2013; Choi and Park, 2015). Furthermore, such changes in cp genomes indicate that they are related to abiotic and/or biotic factors, including life cycle (e.g., parasite vs. nonparasite, Wicke et al., 2013) and habitat (e.g., alpine vs. lowland, Hu et al., 2015). Studies of Asteraceae have served as an excellent opportunity, on a global scale, to understand adaptions that have occurred in the recent radiation of a plant group (Panero and Funk, 2008).
Chloroplast and nuclear DNA analyses indicate that several clades exist within the tribe Gnaphalieae, namely the South African Relhania basal clade (e.g., Relhania L'Hér. and Phagnalon Cass.) and its derived lineages, including Australian clades (Ewartia Beauverd and Euchiton Cass.), the HAP clade (i.e., Helichrysum, AnaphalisDC., and Pseudognaphalium), the FLAG clade (Filago L., Leontopodium R. Br. ex Cass., Antennaria Gaertn., and Gamochaeta Wedd., as well as several small genera), and Gnaphalium L. These clades have consistently been grouped as individual monophyletic lineages even within the crown radiation group, which exhibits very uneven rates of nucleotide substitution and a recent and rapid evolutionary radiation event. Some correlations have been found for the phylogeny and biogeographic patterns of these clades (e.g., taxa of the Australian clade or the distinct distribution range of Helichrysum species). This suggests that more comprehensive knowledge of such correlations is essential if we are to address complex intergeneric relationships.
Asian taxa of the tribe Gnaphalieae, such as Anaphalisand Leontopodium, are regarded as indispensable elements within a transitional lineage extending from the South African basal clade to its derived clades on other continents, even though the tribe has few species (Ward et al., 2009). Because of their position between other clades in a transitional lineage, more detailed information about Anaphalisand Leontopodium is needed for a greater understanding of the phylogeny and evolutionary history of the tribe Gnaphalieae (Ward et al., 2009; Nie et al., 2013). These two genera belong to a distinct lineage and the HAP and FLAG clades occupy nearly half of the crown radiation group. However, previous studies have primarily focused on those clades in more diverse regions, e.g., in Africa and Australia, or on specific genera, such as Helichrysum and Pseudognaphalium. However, more comprehensive phylogenic information is needed with regard to various regions, habitats, and taxonomic positions.
In this study, we investigated the fully sequenced cp genomes of the two representative Anaphalisand Leontopodium species A. sinica Hance and L. leiolepis Nakai. We expect that a comparative analysis of cp genomes from these genera will provide new insight concerning the complex evolutionary histories that extant members of the tribe Gnaphalieae have experienced during their recent, rapid radiation from southern Africa to various new habitats. We believe that this work represents the first comparison of Asian taxa and the cp genomes of other Asteraceae and Gnaphalieae species. Our results will be useful for inferring the inter- and intra-evolutionary histories of the tribe Gnaphalieae, and the genomic information obtained here can be used for efficient propagation of edelweiss species
Materials and Methods
Plant Materials, Genome Amplification, and Sequencing
Fresh, young leaf material of Anaphalissinica and Leontopodium leiolepis were collected from natural populations at Mt. Geumsung and Mt. Seolak in South Korea, and voucher specimens were deposited in the herbarium of Inha University (IUI: Geum014 and 96004, respectively). Total genomic DNA was extracted using a DNeasy Plant Mini Kit (Qiagen, Seoul, Korea), and was pre-washed with STE buffer to remove any inhibitory chemicals (Takakura and Nishio, 2012). The concentration of each extract was measured with a NanoDrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) before high-quality DNA was sequenced using the Illumina Miseq platform (BML, Daejeon, Korea).
Genome Assembly, Annotation, and Analysis
Prior to genome assembly, all raw sequence reads of the two species produced by Illumina were checked by FASTQC (Andrews, 2010) and trimmed by Trimmomatic 0.32 (Bolger et al., 2014). These trimmed raw reads were assembled by a de novo method using Geneious R7 (Drummond et al., 2011). In total, 10,582,958 and 20,246,780 paired-end reads with 282.1 and 283.6 average read lengths were generated for A. sinica and L. leiolepis, respectively. Each cp genome sequence was mapped with reference to that of Artemisia frigida (JX293720), using Geneious R7. To avoid assembly errors from homopolymer runs, and to identify the gaps and borders of the LSC, SSC, and Inverted Repeat (IR) regions, we designed specific primers based on contig sequences and verified those sequences through PCR and Sanger sequencing methods. Each genome was annotated using the online program DOGMA (Wyman et al., 2004), and corrections were made manually for start and stop codons and for intron/exon boundaries. Both trnA and rRNA genes were identified by BLASTN with the same database and by trnAscan- SE (Schattner et al., 2005), with default settings. A circular map of each cp genome was produced by Organellar Genome DRAW (Lohse et al., 2007), followed by manual modifications. The repeating sequences of cpDNA were identified by the SSR Extractor (http:// www.aridolan.com/ssr/ssr.aspx). To compare their synonymous (Ks) and non-synonymous (Ka) nucleotide substitutions, we aligned the functional protein-coding genes of these two Asian taxa using Geneious R7 and analyzed them with DnaSP (Librado and Rozas, 2009). We also used specific primer pairs to verify the pseudogenization (or gene loss) of trnT-GGU with related plant materials, including representatives of the genus category, i.e., Gnaphalium uliginosum; the Australian clade: Euchiton japonicum; members of the FLAG clade: Gamochaeta pensylvanica, L. japonicum, and L. leontopodioides; and members of the HAP clade: A. sinica var. morii, A. margaritacea, Helichrysum cymosum, H. orientale, and Pseudognaphalium affine.
Phylogenetic Analysis
Our phylogenetic analysis covered 13 complete cp DNA sequences that included 11 Asteraceae species and two outgroup species of Apiaceae and Araliaceae that were downloaded from the NCBI database (http://www.ncbi.nlm.nih.gov/genomes). From those species, 52 protein-encoding genes were aligned using MUSCLE (Edgar, 2004), which is part of the Geneious program. We then performed a Maximum Likelihood (ML) analysis that utilized RAxML with the GTR + G model (Stamatakis, 2006; Stamatakis et al., 2008).
Results and Discussion
Characteristics of the Leontopodium leiolepis and Anaphalissinica cp Genomes, and Comparison with Other Asteraceae cp Genomes
The average paired-end read lengths were 301 bp for both L. leiolepis and A. sinica. These were assembled into the reference cp genome sequence of Artemisia frigida, which belongs to the closely related tribe Anthemideae. For L. leiolepis, 662,243 reads were assembled (mean coverage of 1,041.5). After the low-assembly coverage of gap regions was confirmed, we obtained 151,072 bp for the complete cp genome sequence of L. leiolepis (GenBank Accession Number NC027835). Its quadripartite structure includes an LSC region of 83,308 bp, an SSC region of 18,052 bp, and IR regions of 24,856 bp each (Fig. 1). Approximately 59.4% of the L. leiolepis cp genomic sequence (average GC content of 37.3%) is comprised of protein-coding genes, with the remaining 40.6% being non-coding regions such as introns and intergenic spacers. Of the 132 total genes (Table 1), 85 are protein-coding, 37 are for transfer RNA, eight are for ribosomal RNA, and two are partial genes (ycf1 and rps19) located on the boundaries of the IR regions. The IR regions contain seven protein-coding genes, seven trnAs, and four rRNAs that are duplicated. The LSC region contains 62 protein-coding genes and 22 trnA genes, whereas the SSC region has 11 protein-coding genes and one trnA.
For A. sinica, 139,015 reads were assembled (mean coverage of 210). After the low-assembly coverage of gap regions was confirmed, we obtained 152,718 bp for the complete cp genome sequence (GenBank Accession Number KX148081). Its quadripartite structure includes an LSC region of 84,546 bp, an SSC region of 18,488 bp, and IR regions of 24,842 bp each (Fig. 2). Approximately 59.7% of the A. sinica cp genomic sequence (average GC content of 37.1%) is comprised of protein-coding genes, with the remaining 40.3% being non-coding regions such as introns and intergenic spacers. Among the 131 genes (Table 1), 85 are protein-coding, 36 are for transfer RNA, eight are for ribosomal RNA, and two are partial genes (ycf1 and rps19) located on the boundaries of the IR regions, which is similar to the genomic contents of L. leiolepis. The IR regions contain seven protein-coding genes, seven trnAs, and four rRNAs that are duplicated. The LSC region contains 62 protein-coding genes and 21 trnA genes, whereas the SSC region has 11 protein-coding genes and one trnA.
Fig. 1.
Map of the Leontopodium leiolepis chloroplast genome
Among the protein-coding genes of these two cp genomes, 11 have single introns and three (clpP, rps12, and ycf3) have two introns each. In the case of rps12, the 5′-end exon is located in the LSC region, whereas two copies of the 3′-end exon and intron are located in the IR regions. Similar observations have been reported for other Asteraceae cp genomes (Nie et al., 2012; Liu et al., 2013). Our comparisons of nucleotide substitution rates among coding genes revealed that ndhJ had the highest synonymous nucleotide substitution rate (Ks: 0.2777) whereas rpl16 had the highest non-synonymous nucleotide substitution rate (Ka: 0.0238) (Table 2). In contrast, 19 and 32 genes showed no changes in Ks and Ka, respectively.
The cp genomes characterized here did not differ significantly from those of other completed Asteraceae cp genomes. As noted for other angiosperm lineages, the size variation, approximately 2 kb, was primarily attributed to differences in lengths of the LSC region, which ranges from 81.9 kb in Aster spathulifolius to 84.3 kb in Parthenium argentatum (Table 3 and Fig. 3). Other characteristics were similar between these two cp genomes of the tribe Gnaphalieae and those from other members of Asteraceae, including the presence of two pseudogenes, ycf1 and rps19, at the IR boundaries. In fact, previously described cp genomes of Asteraceae have revealed nearly identical gene contents and structures (Timme et al., 2007; Kumar et al., 2009; Dempewolf et al., 2010; Doorduin et al., 2011; Nie et al., 2012; Zhang et al., 2014; Choi and Park, 2015). This is in contrast to the earliest cp-mapping studies, which revealed two large inversions, specifically 22.8 kb and 3.3 kb, in the LSC region (Jansen and Palmer, 1987a, b; Kim et al., 2005). Despite several structural changes, e.g., inversion of the SSC region, being described (Liu et al., 2013; Choi and Park, 2015), those events might simply represent an alternative state in individual plants rather than characteristics unique to a specific lineage. In comparison, the two previously reported large inversions described above demonstrate an ancient evolutionary split within Asteraceae (Jansen and Palmer, 1987a, b; Kim et al., 2005). Indeed, inversion events in the SSC regions may have easily been promoted by their loop structure located between the two IRs. We also found two alternative states for SSC regions that were assembled in exactly the same way except for their direction. Unfortunately, we could not explicitly verify the orientation of the SSC region because our two featured species had short reads (301 bp). However,our assumption is strengthened by a recent report of SSC inversions in Asteraceae (Walker et al., 2015). Therefore, we believe that the events previously described for the SSC region cannot be used to infer the functional evolution among cp genomes and phylogenetic relationships. Moreover, we propose that the cp genomes in the family Asteraceae are remarkably conserved when compared with those of other large plant groups, such Fabaceae or Orchidaceae.
Table 1. Genes identified in the chloroplast genomes of Leontopodium leiolepis and Anaphalissinica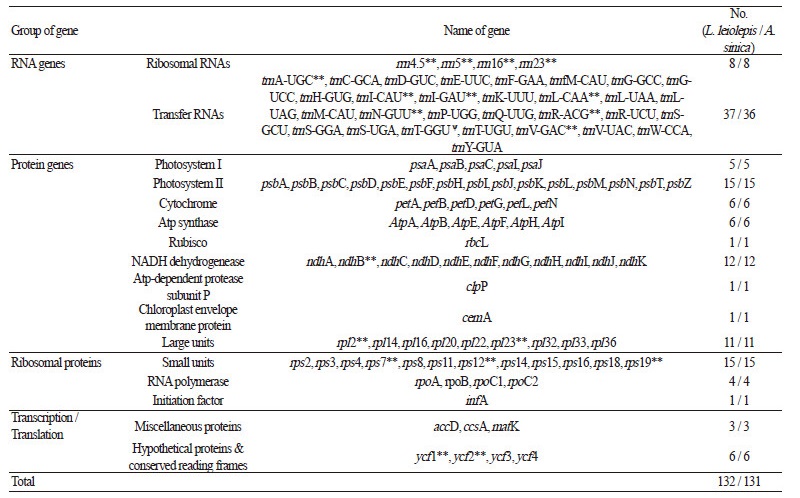 |
Notes : *, genes with introns; **, duplicated genes; ѱ, gene loss in A.sinica |
Fig. 2.
Map of the Anaphalissinica chloroplast genome.
Pseudogenization and Gene Loss of trnT-GGU in Gnaphalieae
The gene contents of the two Gnaphalieae cp genomes are similar to those of other Asteraceae species, with the exception of the gene trnT-GGU. Although trnT-GGU exhibits several base substitutions (1 to 2 bp), it is present and completely functional in all other tribe members in Asteraceae. In comparison, the trnT-GGU gene in L. leiolepis has undoubtedly been pseudogenized, with two indels (6 bp and 8 bp) that can cause a modification to the structure of that trnA. Moreover, we determined that complete trnT-GGU gene loss occurred in the cp genome of A. sinica (Fig. 4).
In addition to large structural rearrangements, relatively small-scale genomic changes, such as gene loss and transformation to pseudogenes, can serve as useful phylogenetic markers when addressing the phylogeny and evolutionary history of land plants. Representative cases have been reported for parasitic plants, including Cuscuta of Convolvulaceae (McNeal et al., 2007), Rhizanthella of Orchidaceae (Delannoy et al., 2011), and genera within Orobanchaceae (Li et al., 2013). These parasites exhibit gene loss or pseudogenization of the majority of chloroplast genes involved in photosynthesis (e.g., atpB, ndhF) because their host plant–dependent life cycle no longer requires autotrophic growth. Although such drastic sequence variation is uncommon in plant cp genomes, such events that reflect phylogeny are increasingly being reported based on the enhanced use of comparative chloroplast sequence analyses (Lee et al., 2007; Schmickl et al., 2009; Do et al., 2014).
Table 2. Comparison of synonymous (Ks) and non-synonymous (Ka) nucleotide substitution rates in the chloroplast genomes of Leontopodium
leiolepis and Anaphalissinica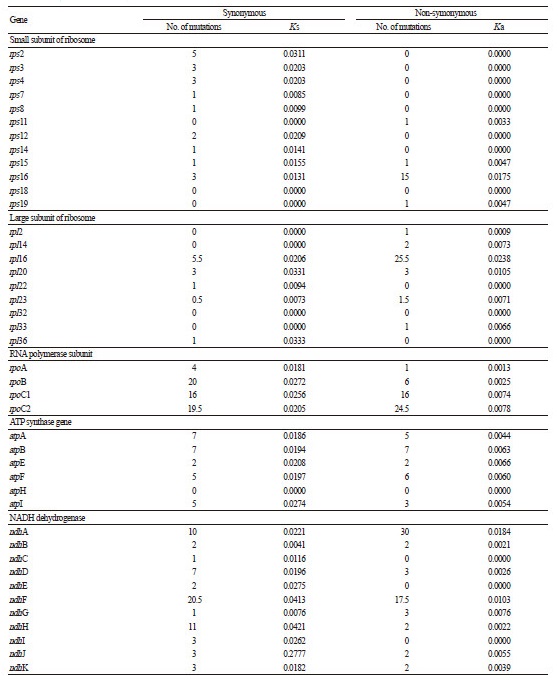 |
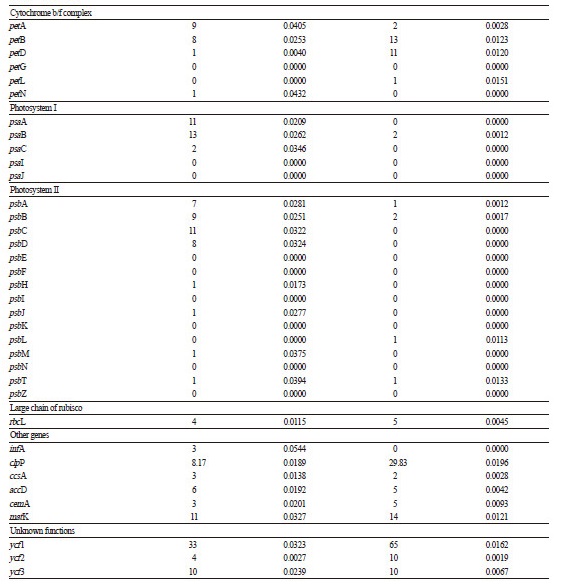
Accordingly, we identified similar features in other genera of the tribe Gnaphalieae. For example, the trnT-GGU genes in the FLAG, Gnaphalium, and Australian clades have been pseudogenized and, with the exception of Pseudognaphalium affine, those in members of the HAP clade have been completely lost. However, we were unable to ascertain whether trnT-GGU pseudogenization or gene loss demonstrates an autapomorphy in the entire tribe due to our limited taxon sampling size. Nevertheless, the near consistent trnT-GGU mutations clearly contrast with the gene contents for other tribes in Asteraceae.
Table 3. Comparison of sequence characteristics among nine chloroplast genomes of Asteraceae species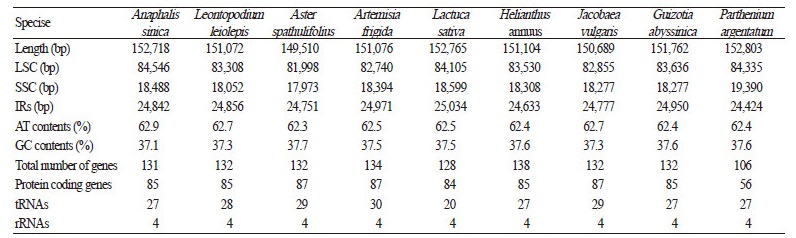 |
Fig. 3.
Comparison of Inverted Repeats (IRs) and chloroplast genome structure among Asteraceae species.
Therefore, the trnT-GGU gene sequence may be a useful phylogenetic marker for investigating the complicated phylogeny and evolutionary history of the tribe Gnaphalieae. In particular, pseudogenized trnT-GGU in P. affine and complete trnT-GGU gene loss in Helichrysum and Anaphaliswithin the HAP clade indicate their evolutionary history. In general, members in the HAP clade are thought to have originated via allopolyploidy. This conclusion is robustly supported by various cytological, morphological, and molecular features (Smissen et al., 2011; Galbany-Casals et al., 2014). Phylogenetic analyses of Helichrysum and related genera have provided further proof of such origins, even though the potential parental species are not certain. Nevertheless, our current results give additional information about their lineage. If one considers the maternal inheritance and highly conserved structures of cpDNA, then the presence of the pseudogenized trnT-GGU in P. affine, unlike that in other members of the HAP clade, suggests that neither Helichrysum nor Anaphalis, which both exhibit complete trnT-GGU gene loss, form the maternal lineage for the genus Pseudognaphalium. Instead, one of the genera within the FLAG clade, which exhibits a pseudogenized version of trnT-GGU, would be a more appropriate maternal lineage candidate. Likewise, we can hypothesize that the genus Anaphalis(x = 14), which features complete trnT-GGU gene loss, originated by allopolyploidy between Helichrysum (x = 7) as a maternal lineage and Pseudognaphalium (x = 7) as a paternal lineage. Consequently, our cpDNA data, including gene contents and structure, serve as useful phylogenetic markers for studies of the phylogeny and evolutionary history of the tribe Gnaphalieae. Examination of further members of the tribe Gnaphalieae will facilitate a more comprehensive understanding of its dynamic evolutionary history.
Fig. 4.
Comparison of trnT-GGU sequences among Asteraceae species.
Phylogenetic Analysis of Asteraceae cp Genomes
We used 52 protein-coding genes to generate a phylogenetic reconstruction of Asteraceae. This analysis included the cp genomes of all examined species alongside two Apiales species, namely Anthriscus cerefolium and Kalopanax septemlobus, as outgroup taxa. Phylogenetic analysis produced three clades that corresponded to the classification system of Carduoideae, Cichorioideae, and Asteroideae. Within the Asteraceae, Centaurea diffusa of Carduoideae was located at the lowest level whereas Lactuca sativa of Cichorioideae formed a sister group with species in Asteroideae (Fig. 5). The subfamily Asteroideae formed two groups: Senecionodae/Helianthodae and Asterodae. Following the incorporation of further protein-coding genes into this reconstruction (data not shown), we were able to group Jacobaea vulgaris of Senecinodae with the Asterodae. However, such a phylogenic relationship may be the result of biased topology due to a lack of gene annotations for some taxa. Within Asterodae, we found that the monophyletic group of A. sinica and L. leiolepis was a sister group to other species within Astereae (Aster spathulifolius) and Anthemideae (Chrysanthemum indicum, Artemisia frigida, and A. montana). The topologies defined here are very consistent with previously reported phylogenetic relationships based on chloroplast genes (Panero and Funk, 2008). Moreover, our finding of a phylogenetic relationship for two Asian genera in Gnaphalieae further supports our conclusion that the tribe Gnaphalieae, albeit independent, is closely related to Anthemideae and Astereae rather than to the tribe Inuleae in Helianthodae, which was previously regarded as a sister group.
Fig. 5.
Phylogenetic analysis using 52 chloroplast protein genes and Maximum Likelihood method
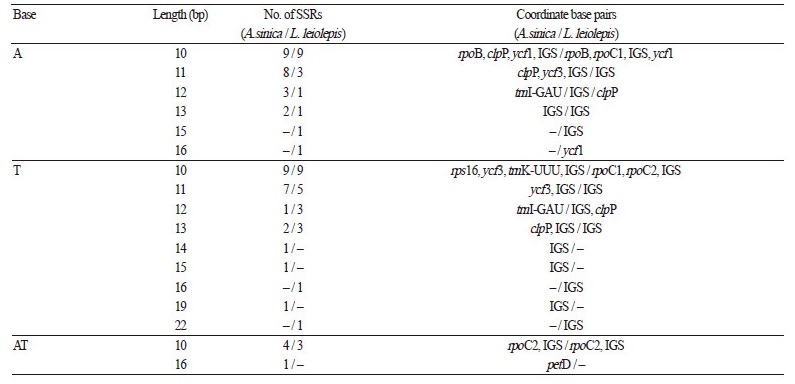
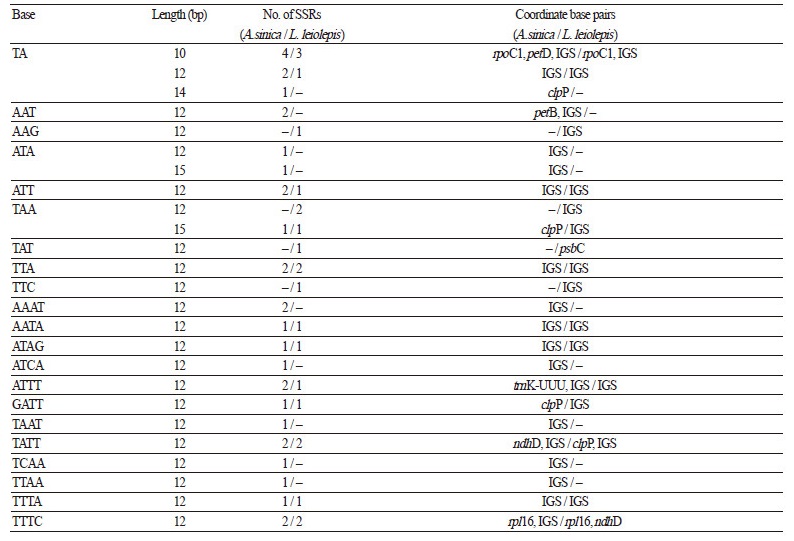
Repeat Sequence Analysis
Due to the high-level polymorphism that exists even among plants of the same species, microsatellites and simple sequence repeats (SSRs) are widely used as valuable genetic markers for assessing population genetic diversity, especially in studies of conservation genetics for endangered and rare species (Kramer and Havens, 2009). L. leiolepis and its relatives, commonly known as “edelweiss”, are primarily found in the upper regions of high mountains and are considered as endangered species. Thus, a number of edelweiss species undoubtedly require suitable management strategies in many countries. Here, we found 63 and 81 SSRs, including 38 and 44 mono-, 7 and 12 di-, 9 and 9 tri-, and 9 and 16 tetranucleotides in the cp genomes of L. leiolepis and A. sinica, respectively (Table 4). The mononucleotide repeats exclusively contained the nucleotide A (n = 16 and 22, respectively) or T (n = 22 and 22, respectively), similar to that in other angiosperm species. This A/T-rich tendency was consistent throughout almost all SSR nucleotide categories. These SSR data provide a powerful tool for evaluating the population genetics of these plant species, and will help to make informed decisions about suitable conservation management strategies.
In conclusion, this is the first study to analyze the complete chloroplast genomes of two species in the tribe Gnaphalieae within Asteraceae, namely those of A. sinica and L. leiolepis. Their cp genome characteristics, such as copy number and gene content/order, closely resemble that of previously reported cp genomes of other Asteraceae species. However, the observed pseudogenization or gene loss of trnT-GGU in these studied species reinforces the taxonomic boundary of the tribe Gnaphalieae as an independent group, and can be used as an informative molecular marker for further research into generic and/or tribal relationships. Finally, the SSRs identified here provide valuable species–specific genetic information that may assist in conservation efforts.