

Introduction
Materials and Methods
Sample Preparation
Nanopore Library Preparation and Sequencing
Metagenomic Analysis
Phylogenetic Analysis
Data Availability
Transmission Electron Microscopy (TEM)
Pathogenicity Test and Validation of CMV by RT-PCR
Results
Identification of CMV in Mint by Nanopore Sequencing
Determination and Characterization of the Genome Sequence of CMV Isolates
Phylogenetic Analyses
CMV Infection and Detection in N. benthamiana
Discussion
Introduction
Mints (Mentha spp.) have been cultivated for centuries due to their unique fragrances, medicinal properties, and nutritional benefits. Mints include more than 45 species, including M. arvensis L. (Corn Mint), M. spicata L. (spearmint), and M. x piperita (peppermint), and are cultivated worldwide (Tzanetakis et al., 2006). As demand increases, the production of mint plants has been rising annually in South Korea (Baek et al., 2016).
Mint plants are propagated clonally, and mint viruses have traveled with repeatedly propagated clones around the world (Tzanetakis et al., 2006). Several plant viruses, including the arabis mosaic virus, alfalfa mosaic virus (AMV), strawberry latent ringspot virus, tobacco ringspot virus, cucumber mosaic virus (CMV), tomato aspermy virus, mint vein banding associated virus, mint virus-1, mint virus-2, peppermint stunt virus, tomato spotted wilt virus, and mint virus X, have caused serious economic losses by damaging Mentha species (Tzanetakis et al., 2006). These mint viruses are also transmitted by a wide range of organisms including nematodes, aphids, thrips, and whitefly. Infection with a single virus or a mixture of multiple viruses induces yellow vein, leaf yellowing, mosaic, crinkling, and cupping symptoms (Tzanetakis et al., 2006; Jeong et al., 2021). Nucleic acid amplification assays such as the reverse-transcription assay followed by the polymerase chain reaction (RT-PCR) technique are commonly used to identify mint viruses (Tzanetakis et al., 2006; Saeed et al., 2014; Menzel and Winter, 2017; Heo and Choi, 2021). Recently, high-throughput sequencing (HTS) studies using Illumina sequencing platforms have been conducted to identify AMV in M. haplocalyx (Zhengnan et al., 2019). State-of-the-art diagnostic platforms for plant virus detection, as a next generation biosecurity management approach, can afford rapid, efficient, and accurate identification and reliable management of viral infections (Van der Want and Dijkstra, 2006; Aboul-Ata et al., 2011; Vazquez-Iglesias et al., 2022).
Oxford Nanopore Technologies (ONT) provides third-generation sequencing platforms that facilitate real-time single-molecule DNA or RNA sequencing, methods capable of directly identifying DNA and RNA molecules through nanopore proteins embedded in a polymer membrane (Eid et al., 2009; Deamer et al., 2016; Phannareth et al., 2020). ONT sequencing platforms overcome the limitations of other HTS technologies by combining membrane-based sequencing with personal- computer-based metagenomic analyses in the field as well as in the laboratory (Theuns et al., 2018). ONT nanopore sequencing has several advantages, including long reads, a short sequencing time, and real-time interpretation of sequencing results (Batovska et al., 2017). For virus diagnostics, the MinION sequencer and Flongle (flow cell dongle) are simple and powerful platforms, providing reliable sequencing data in a short time, i.e., within 48 h (Vazquez-Iglesias et al., 2022).
In this study, we report a method for the rapid identification of viruses that infect spearmint plants by a virome analysis using an ONT MinION sequencing platform.
Materials and Methods
Sample Preparation
In August of 2022, spearmint (M. spicata) plants with disease foliar symptoms of chlorosis and mosaic (Fig. 1) were collected from Gwangju, Korea, and symptomatic leaf samples were immediately stored at ‒80 °C. Total RNA was extracted from the symptomatic leaf samples (‒200 mg) using a Clear-S™ Total RNA extraction kit (InVirusTech Co., Gwangju, Korea) according to the manufacturer’s instructions. DNA in the extracted total RNA samples was completely removed using a RapidOut DNA Removal Kit (Thermo Fisher Scientific, Waltham, MA, USA). The quality of the extracted RNA was determined using a Qubit™ 4 Fluorometer, a Qubit™ RNA HS Assay Kit (Thermo Fisher Scientific), and a spectrophotometer (BioDrop, Biochrom Ltd., Cambridge, UK). Plant rRNA molecules were further removed using a QIAseq FastSelect -rRNA Plant Kit (Qiagen, Hilden, Germany) to obtain enriched viral RNA (ribodepletion). The RNA was then purified with the AMPure XP Reagent (Beckman Coulter, Brea CA, USA) and quantified with a Qubit™ 4 Fluorometer and a Qubit™ RNA HS Assay Kit.
Nanopore Library Preparation and Sequencing
The qualified RNA was used to construct a library with a direct cDNA sequencing kit (SQK-DCS109) with a Flongle Starter pack (Oxford Nanopore Technologies, Oxford, UK), as described by Lee et al. (2022). Briefly, cDNA was synthesized using a Maxima H Minus Double-Stranded cDNA Synthesis Kit (Thermo Fisher Scientific). Then, dA-tailing was conducted using a NEBNext® Ultra™ II End Repair/dA-Tailing Module (New England Biolabs, Ipswich, MA, USA) with double-stranded cDNA, with adapter ligation done using a Blunt/TA Ligase Master Mix (New England Biolabs) with Adapter Mix (SQK-DCS109; Oxford Nanopore Technologies). The final concentration of the nanopore library was 3.3 ng/µL. The prepared nanopore library was loaded into the Flongle flow cell (R9.4.1) port of the MinION (Oxford Nanopore Technologies), and sequencing was conducted for 24 hours through the MinKNOW software (version 21.11.7). The reads were analyzed in real time using the high-accuracy mode (quality score of 7) with Guppy software (version 5.1.12) and the MinKNOW software.
Metagenomic Analysis
The FASTQ files generated were converted to FASTA files, and the adapter sequences were removed using Geneious Prime (version 21.1.1). Identification of the sequences of the plant viruses was done through the ONT EPI2ME WIMP workflow (version 3.4.2) and with a BLASTn search (-outfmt 6) of the sequences using National Center for Biotechnology Information (NCBI) Viral Genome data (Viral RefSeq Database; VirDB; version 21.11.4). The virus-associated reads were mapped to plant virus genomes that scored highly in NCBI BLASTn searches on Geneious Prime, and the consensus sequences were obtained from alignments with the requirement of a minimum of coverage depth of 1x and a support fraction of at least 20% for base-call ambiguity.
Phylogenetic Analysis
Multiple sequence alignments were performed using the ClustalW Multiple alignment tool of MEGAX (version 10.2.4) on the plant virus genome sequences obtained in this study and the viral genome sequences from NCBI BLASTn. The phylogenetic analysis was constructed here used the CMV-CNU-spearmint isolate (LC744757, South Korea, Mentha spicata) and 17 other CMV isolates (AM183119; CMV-Ri-8; Spain; Solanum lycopersicum, GU327365; CMV-Rb; South Korea; Rudbeckia hirta, D10538; CMV-Fny; USA, D00385; CMV-O, D12499; CMV-Y; Japan; AB004781; CMV-D8; Japan, EF216867; CMV-Cb7; China; Solanum lycopersicum, AY429432; CMV-Ca; China; Arachis hypogaea, AB042294; CMV-IA; Japan, EF213025; CMV-CTL; China; Brassica chinensis, U20219; CMV-Ix; USA; Lycopersicon esculentum, M21464; CMV-Q, AF198103; CMV-LY; Australia, EF202597; CMV-Tsh; China; Lycopersicon esculentum, L15336; CMV-Trk7, AF063610; CMV-S; South Africa, AB176847; CMV-TN; Japan; Nicotiana tabacum) from GenBank. Neighbor-joining (NJ) and maximum likelihood phylogenetic trees were generated based on coat protein gene sequences with 1000 bootstrap replicates, and the Tamura-Nei model was used for nucleotide substitution as determined by MEGAX (version 10.2.4) (Tamura et al., 2004; Kumar et al., 2018).
Data Availability
Sequencing data files in the FASTQ format were deposited in the NCBI Sequence Read Archive (SRA) repository under SRR22891755, and the viral genome sequences obtained were deposited in GenBank with accession numbers.
Transmission Electron Microscopy (TEM)
Spearmint (M. spicata) leaf tissues with viral infection-like symptoms and healthy leaf tissues were examined by TEM. Negative staining was conducted on copper grids (200-mesh) while fixation, while dehydration, and infiltration processes were performed. Crude sap from the leaf samples (5 µL) was placed on the 200 Hex grid for 5 min (EMS Inc., Hatfield, PA, USA). The grids were washed twice with distilled water for 30 s and incubated in a solution of 1.5% uranyl formate (EMS inc.). The grids were then air-dried and imaged by a JEM-2100F TEM system (JEOL Ltd., Tokyo, Japan).
Pathogenicity Test and Validation of CMV by RT-PCR
Nicotiana benthamiana plants were inoculated with symptomatic mint leaf sap in a 0.5 M NaPO4 buffer (pH 7.2). The inoculated plants were grown in a constant-temperature growth chamber at 24°C with a 14/10 h (light/dark) photoperiod. The presence of the virus identified by ONT sequencing in spearmint plants was confirmed by RT-PCR using previously reported diagnostic primers (Choi et al., 1999).
Results
Identification of CMV in Mint by Nanopore Sequencing
For the rapid identification of the virus infecting the mint plants, ONT MinKNOW software was utilized to analyze mint tissues showing viral infection-like disease symptoms (Fig. 1). The sequencing generated 347,241 raw reads with an average read length of 404.6 nucleotides. In total, 42,229 reads with an average read length of 110.3 were obtained after adapter trimming and quality filtering steps (>Q7) (Table 1). Following the WIMP workflow, 35,800 (10.3%) virus-like reads were identified. To identify viral genomes that match the reads, trimmed reads were BLASTed on the NCBI VirDB. Most of these virus-like reads were aligned with CMV (RNA 1, 698 reads; RNA 2, 4092 reads; and RNA 3, 2624 reads) (Table 2).

Table 1.
Summary of ONT sequencing data
Table 2.
List of identified viruses in spearmint samples
Determination and Characterization of the Genome Sequence of CMV Isolates
De novo genome assembly of the MinION reads was conducted on the viral reference genomes of CMV isolates derived from GenBank (Fig. 2). For CMV RNA 1, a total of 1287 reads (0.4% of all reads) were mapped with 92.9% query coverage and 92.8% nucleotide identity. For CMV RNA 2, a total of 1290 reads (0.4% of all reads) were mapped with 97.3% query coverage and 93.1% nucleotide identity. In the case of CMV RNA 3, a total of 2324 reads (0.7% of all reads) were mapped with 96% query coverage and 93.6% nucleotide identity. Nearly complete RNA 1, RNA 2, and RNA 3 genome consensus sequences of the CMV infecting spearmint (CMV-CNU-spearmint) were deposited in GenBank (RNA 1, LC744755; RNA 2, LC744756; RNA 3, LC744757).

RNA 1, RNA 2, and RNA 3 of CMV-CNU-spearmint consisted of 3120, 2968, and 2128 nucleotides, respectively, exhibiting a genome structure similar to those of other CMV isolates reported previously (Nouri et al., 2014). RNA 1 contained an ORF (ORF1a) of 991 amino acids; RNA 2 contained two ORFs of 857 aa (ORF2a) and 111 (ORF2b) aa; and RNA 3 contained two ORFs of 280 (ORF3a) and 219 aa (ORF3b). CMV-CNU-spearmint shared nucleotide sequence similarities at 91.5–98.8% for ORF1a, 96.4–99.2% for ORF2a, 96.1–98.8% for ORF2b, 97.7–99.7% for ORF3a, and 97.5–99.5% for ORF3b with other isolates. The corresponding amino acid sequence similarity rates were 97.1–99.7% for ORF1a, 97.6–99.1% for ORF2a, 93.6–98.1% for ORF2b, 97.4–100% for ORF3a, and 98.1–100% for ORF3b.
Phylogenetic Analyses
To determine the relationship between the CMV-CNU-spearmint isolate and the 17 other CMV isolates from GenBank mentioned above, the complete CP (ORF3b) nucleotide and amino acid sequences were analyzed and phylogenetic trees were constructed using the Maximum Likelihood algorithm (Fig. 3). The phylogenetic analysis revealed that the isolates formed well-separated clusters in subgroups IA, IB, and II. The CMV-CNU-spearmint isolate was clustered in the IA subgroup with five other CMV isolates: Spain (CMV-Ri-8), USA (CMV-Fny), South Korea (CMV-Rb), Japan (CMV-Y and CMV-D8), and CMV-O.
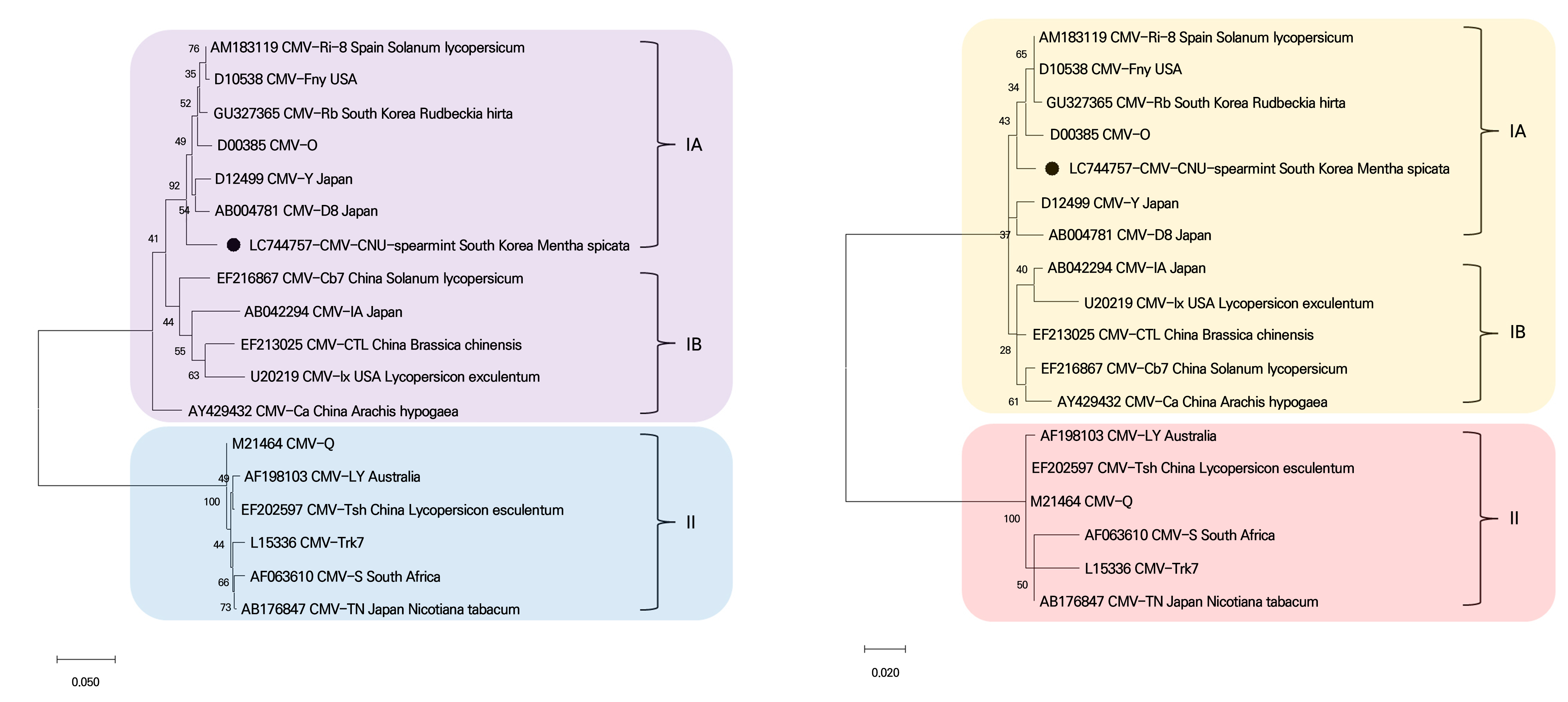
Fig. 3.
Phylogenetic analysis using the Maximum Likelihood algorithm based on complete coat protein nucleotide (A) and amino acid (B) sequences of CMV isolates. For each molecular group (IA, IB, and II), the reference sequences are indicated with their accession numbers and names. The genome of the CMV-CNU-spearmint isolate is indicated by the black filled circles. The scale bar represents the genetic distance.
CMV Infection and Detection in N. benthamiana
To confirm the infectivity of the CMV-CNU-spearmint isolate, sap from frozen mint samples was used to inoculate Nicotiana benthamiana plants mechanically. N. benthamiana inoculated with symptomatic spearmint leaves exhibited severe mosaic chlorosis and leaf curling 30 days post-inoculation (Fig. 4A). Transmission electron microscopy revealed icosahedral virions with diameters of about 30 nm on symptomatic leaves (Fig. 4B). CMV-CNU-spearmint infection in N. benthamiana plants was confirmed by RT-PCR using CMV-specific diagnostic primers (Choi et al., 1999). Amplicons of the expected size (938 bp) were obtained from three symptomatic samples (Fig. 4C). These amplicons were further confirmed by Sanger sequencing (data not shown).

Fig. 4.
Pathogenicity test of CMV-CNU-spearmint in N. benthamiana plants. (A) Visible symptoms of symptomatic spearmint leaves of inoculated N. benthamiana plants at 30 days post-inoculation. (B) Transmission electron microscopy image of negatively stained CMV-CNU-spearmint particles. TEM magnification range: ×135,000. (C) Detection of CMV-CNU-spearmint in infected N. benthamiana plants by RT-PCR. M, 100-bp ladder; Lane 1, CMV-spearmint-inoculated sample; Lane 2, mock-inoculated sample.
Discussion
Despite the increasing importance of mint plants worldwide, the viruses infecting mint in Korea have not been identified to date. In this study, CMV was identified as the causal agent of disease in spearmint plants in Gwangju, South Korea, using the ONT sequencing platform. The ONT platform has been successfully utilized to identify viruses present on crops through a metagenomics-based, rapid, reliable, real-time, and portable approach (Filloux et al., 2018). This study is the first report of a natural CMV infection in spearmint in Korea. CMV is one of the most destructive viruses, affecting more than 1300 plant species worldwide. More importantly, the number of host plants continues to increase (Akhtar et al., 2019), and the list of potential natural hosts of CMV in Korea is expanding (Lee et al., 2020; Bae et al., 2021; Nam et al., 2022).
In this study, the genomic reads generated by the ONT-based sequencing platform were almost completely assembled into the CMV reference genomes (RNA 1, RNA 2, and RNA 3). The identified CMV isolate was found to be closely related to Asian CMV isolates under the subgroup IA. Currently, the greatest shortcoming of ONT-based sequencing is its generation of a low number of reads and its reduced accuracy compared to short-read sequencers (Sun et al., 2022). This study also showed that the genomic sequence of CMV-CNU-spearmint as identified here was incompletely assembled at the 3’-UTRs. However, these limitations do not have a major impact on virus identification (Sun et al., 2022). The identified CMV isolate was further examined biologically by pathological tests, in this case TEM for virion observation and RT-PCR using specific diagnostic primers, in N. benthamiana plants inoculated with the CMV isolate. CMV was also found to be the causal viral agent in mint plants in European countries (Tzanetakis et al., 2010).
In conclusion, the ONT platform was successfully utilized to identify a previously unknown virus presently infecting spearmint plants in Korea. Given the growing amount of data generated by this technology, ONT-based sequencing will be a promising approach for the rapid identification of viral infections in other crops, contributing to the management of plant viral diseases. To the best of our knowledge, this is the first report of a CMV infection in spearmint in South Korea. Further surveys of CMV infections in spearmint as well as other Mentha spp. plants are required for sustainable and effective disease management to avoid further spreading of these viruses.
