

서 언
재료 및 방법
유전자 재동정
도메인 구조 분석
MADS 도메인의 보존된 아미노산 서열 분석
GO term 분석
모티프 분석
계통수 분석
CAFE 분석
P. pyrifolia MADS-box 유전자의 전사체 분석
결과 및 고찰
사과 및 배에서의 MADS-box 유전자군 재동정 및 특징 분석
MADS-box 유전자의 계통수와 CAFE 분석
P. pyrifolia에서 저온 스트레스에 따른 MADS-box 유전자의 발현 및 기능 분석
서 언
전사인자(Transcription factor, TF)는 식물에서 유전자 발현을 조절하는 데 필수적인 역할을 하며, promoter와 enhancer 부위에 결합하여 기본적인 전사 조절, 발달, 세포 신호전달, 환경 반응, 세포 주기 조절 등 다양한 생리적 과정을 수행한다(Moon et al. 2014). 이 중에서도 식물 발달과 환경 반응에 관여하는 MADS-box 유전자는 다양한 기능을 갖는 전사인자군이다. MADS-box 유전자들은 주로 큰 유전자군을 이루고 있으며, 이들은 Serum response factor(SRF) 도메인을 포함하는 전사인자로 구성되어 있다(Wang et al. 2015). MADS-box 전사인자의 구조는 MIKC-type의 경우 MADS(M), Intervening(I), Keratin-like(K), C-terminal(C) 도메인(domain)으로 나뉜다(Theissen et al. 2000). MIKC-type 구조는 M 도메인이 DNA 결합, I 도메인이 단백질 상호작용, K 도메인이 이합체 형성, C 도메인이 전사 활성화를 담당하여 식물 발달을 조절하는 데 중요한 역할을 한다.
사과(Malus domestica)와 배(Pyrus pyrifolia)는 한국에서 가장 많이 재배되는 과일로, 각각 전체 과일 생산액의 24%와 10%를 차지하고 있어 경제적으로도 중요한 가치를 지닌 작물로서, 품종 개선 및 생장 발달 관련 연구가 활발히 이루어지고 있다(Lee et al. 2023; KREI 2024). 사과와 배에서 MADS-box 유전자는 과수 생리학 및 농업적 특성에 중요한 역할을 한다고 알려져 있다. 예를 들어, 사과의 MADS-box 유전자 중 MdJOINTLESS는 과실 낙과와 관련이 있으며, 이 유전자의 발현 분석이 농업적으로 중요한 가치를 지닌다고 알려져 있다(Kim et al. 2012). 또한, 배에서 MADS-box 유전자는 과실 발달과 숙성에 중요한 역할을 수행하는 것으로 보고되고 있다(Ubi et al. 2013).
하지만 두 작물의 MADS-box 유전자에 대한 비교 분석 및 기능적 연구는 여전히 초기 단계에 머물러 있으며, 특히 저온과 같은 비생물적 스트레스에 대한 MADS-box 유전자의 반응 메커니즘은 충분히 연구되지 않았다. 본 연구는 이러한 간극을 메우기 위해 사과와 배의 MADS-box 유전자를 재동정하고 비교 분석하였다. 우리는 재동정된 MADS-box 유전자의 구조적 특성과 진화적 관계를 규명하고, 저온 스트레스 조건에서 이들 유전자의 발현 패턴을 분석하여 주요 후보 유전자를 식별하였다. 마지막으로 Co-expression 네트워크 분석을 통해 저온 저항성을 향상시킬 수 있는 핵심 유전자를 선정하였다. 이러한 연구는 사과와 배의 품종 개선에 필요한 기초 자료를 제공하고, 비생물적 스트레스 적응성을 강화하는 새로운 육종 전략을 제시하는 데 기여할 것으로 기대된다.
재료 및 방법
유전자 재동정
사과와 배에서 MADS-box 유전자를 재동정하기 위해 NCBI(https://www.ncbi.nlm.nih.gov/)에서 Malus domestica, Pyrus pyrifolia의 유전체 데이터를 수집하였다. 그리고 이후 분석을 위한 모델 식물로 Arabidopsis thaliana, Oryza japonica의 유전체 데이터를 수집하였다. 재동정 과정에는 TGFam-Finder v1.20이 사용되었다(Kim et al. 2020). InterProScan 5(-f tsv -appl Pfam)(Jones et al. 2014)로 생성한 TSV 파일을 도메인 식별 파일로 사용하였고, 타겟 도메인 아이디는 PFAM 데이터베이스에서 SRF-domain에 해당하는 PF00319으로 설정하였다. 이때 사용된 매개 변수는 ‘EXTENSION_LENGTH’ = 100,000, ‘MAX_INTRON_LENGTH’ = 100,000, ‘HMM_CUTOFF’ = 1e-3이다. 재동정 과정에서 식별된 유전자에는 새로운 이름이 부여되었다.
도메인 구조 분석
4종의 유전체 데이터 중 펩타이드 파일과 TSV 파일을 사용하여 도메인 구조를 분석하였다. 이 과정에서 분석의 정확도를 높이기 위해 TSV 파일에서 도메인의 e-value가 1e-4 이상인 유전자를 찾아 제거하는 작업을 진행하였다. 그리고 4종의 MADS-box 유전자에서 각 도메인의 개수를 확인하였다. 선행 연구에서의 MADS-box 유전자의 type 분류 기준에 따라 분석을 진행하기 위해, K-box domain을 가진 유전자와 가지지 않은 유전자로 분류하여 각각의 개수를 파악하였다(Masiero et al. 2011).
MADS 도메인의 보존된 아미노산 서열 분석
MADS 도메인의 아미노산 서열은 4종의 식물 유전체에서 추출하였다. MADS 도메인 서열을 정렬하기 위해 MAFFT v7.470(--reorder --maxiterate 1000)(Katoh and Standley 2013)을 사용한 후, TrimAL v1.4(Capella-Gutiérrez et al. 2009)의 -gt 0.1 매개변수를 적용하여 정렬된 서열을 다듬었다. 모든 MADS 도메인의 아미노산 서열 구성을 시각화하기 위해 WebLogo v2.8.2(Crooks et al. 2004)를 사용하였다. 각 종에서의 MADS 도메인 서열을 분석하기 위해 EMBOSS Cons를 plurality 0.1 옵션으로 사용하였다(https://www.ebi.ac.uk/jdispatcher/msa/emboss_cons?stype=protein). 또한, MADS 도메인의 이차 구조를 예측하기 위해 Jpred(Drozdetskiy et al. 2015)를 이용하여 다중 서열 정렬을 수행하였다. MADS 도메인의 α-나선(α-helix)과 β-시트(β-sheet) 구조를 기반으로 MADS 도메인을 7개 영역으로 나눈 후, 각 영역에 대한 서열 보존 점수를 계산하였다. 각 구획의 평균 점수는 Jalview v2.11.4.1을 사용하여 계산하였다(Waterhouse et al. 2009).
GO term 분석
우선, interproscan-5.22-61.0을 통해 MADS-box 유전자 기능 분석을 위해 앞서 탐색한 유전자의 기능적 도메인을 기반으로 GO term을 할당하였다. 이후 OmicsBox 3.4.5(https://www.biobam.com/omicsbox/)를 사용하여 subfamily에서 GO category별로 GO term의 기능적 풍부도를 평가하였다. 이 분석에서는 MADS-box 아미노산 서열에 초점을 맞추었고, BLASTP를 사용하여 NCBI non-redundant proteins database(nr v5)와 비교 및 정렬을 수행하였다. 이때 e-value < 1e-3로 설정되었다. BLAST 결과를 이용하여 해당 GO term으로 mapping과 annotation을 추가로 수행하였으며, 이 과정에서 기본 매개변수를 사용하여 분석을 진행하였다. 분석은 생물학적 과정(Biological Process, BP), 분자 기능(Molecular Function, MF), 세포 구성(Cellular Component, CC)의 세 가지 GO 카테고리로 나누어 유의미한 상위 다섯 기능의 GO term을 선별하였다. 최종적으로 도출된 GO term은 subfamily 간의 기능적 차이를 해석하는 데 활용하였다.
모티프 분석
앞서 e-value cut을 진행한 펩타이드 파일에서 MADS-box 유전자의 도메인에 해당하는 서열 영역만 도출하여 새로운 펩타이드 파일을 만들었다. 이렇게 만들어진 펩타이드 파일로 MEME v5.1.1을 사용하여 MADS-box 유전자의 도메인에서 어떤 모티프가 가장 잘 보존되었는지 확인하였다(Bailey et al. 2006). 이때의 매개 변수는 -protein -oc result -V -time 18000 -mod zoops -nmotifs 50 -minw 10 -maxw 50 -objfun se -markov_order 0이다. 또한 MAST v5.1.1을 이용하여 확인된 모티프의 위치를 추정하였다(Bailey and Gribskov 1998). MEME 결과에서 LLR(Log Likelihood Ratio) 값이 낮은 모티프는 실제로는 존재하지 않을 가능성이 높으므로 LLR 값이 낮은 모티프들은 투명하게 표현되었으며, 이러한 모티프는 신뢰성이 낮다고 판단되어 분석에서 제외하였다.
계통수 분석
우리는 먼저 이전 연구에 설명된 방법을 따라서 계통발생 분석을 수행했다(Jang et al. 2024). 간단하게 설명하면 애기장대, 벼, 사과, 배에서 470개의 재동정된 MADS-box 유전자 펩타이드 서열들을 MAFFT v7.470을 사용하여 정렬하였다(Katoh and Standley 2013). 그 후 trimAl v1.4(-gt 0.1)을 통해 부정확한 서열을 제거하였다(Capella-Gutiérrez et al. 2009). 유전자 간의 진화적 관계를 알아보기 위해 IQ-TREE v2.0.6을 사용하여 유전자 간의 진화적 관계를 조사하고, iTOL v6를 이용하여 이를 시각화하였다(Bui et al. 2020; Letunic and Bork 2021). 추가적인 분석을 위해 A. thaliana와 M. domestica의 선행연구와 계통수에서 보이는 계통적 관계를 기준으로 유전자들의 subfamily를 나누었다(Kumar et al. 2016).
CAFE 분석
Subfamily 별 유전자 수의 확장을 분석하기 위해 CAFE v5를 통한 분석을 진행하였다(Mendes et al. 2021). CAFE 입력 데이터에서는 MADS-box subfamily를 orthologous groups로 간주하여, 각 종이 속하는 분류학적 계통 내에서 subfamily 유전자 수의 확장을 분석할 수 있도록 하였다. Timetree(Kumar et al. 2022)에서 4종에 대한 계통 정보를 추출하고 이를 기반으로 각 subfamily에서 유전자수의 증감을 파악하여 subfamily의 확장을 iTOL v6(Letunic and Bork 2021)을 통해 시각화 하였다.
P. pyrifolia MADS-box 유전자의 전사체 분석
우리는 저온 스트레스 조건에서 P. pyrifolia의 RNA sequencing(RNA-seq) 데이터를 조사하여 MADS-box 유전자의 발현 프로파일을 얻었다(SRX7113732, SRX7113731, SRX7113730, SRX7113729, SRX7113728, SRX7113727, SRX7113726, SRX7113725, SRX7113724, SRX7113723, SRX7113722, SRX7113721 in P. pyrifolia). P. pryifolia의 RNA-seq 데이터는 저온 조건(4°C)에서 0, 15, 30, 45일에 걸쳐 생성되었다. 최초 FASTQ 파일은 CLC Assembly Cell(CLC, Bio, Aarhus, Denmark)로 trimming 하였다. 리드를 매핑한 후, 전체 유전자의 Fragment Per Kilobase per Million mapped reads(FPKM) 값을 StringTie(-e -B -G)를 사용하여 계산하였다(Pertea et al. 2015). 재동정 과정에서 새로운 이름이 부여된 유전자들의 아이디를 원래의 locus 아이디로 변경하였고, MADS 유전자들의 데이터와 전체 유전자들의 데이터를 병합하여 gtf 파일로 생성하였다. Python 스크립트(prepDE.py3)를 실행하여 FPKM 값을 read count로 변환하였고, P. pryifolia에서 log2FoldChange > 2 또는 < ‒2, 그리고 조정된 p-value < 0.05를 기준으로 차등 발현 유전자(DEGs)를 식별하였다(Love et al. 2014).
또한, 우리는 P. pryifolia에서 DEGs와 전체 MADS-box 유전자의 발현 기반 클러스터링 분석을 수행하였다. 이는 이전 연구들에서 목표 유전자와 DEGs 간의 발현 패턴 분석을 통해 목표 유전자의 기능을 추론한 방법을 따랐다(Shen et al. 2020; Feng et al. 2022). R의 Mfuzz 패키지를 사용하여 P. pryifolia의 MADS-box 유전자와 DEGs를 저온 스트레스 하에서 다양한 시간대의 발현 패턴을 기반으로 그룹화하였다(Kumar and Futschik 2007). 그룹화에는 k-average 알고리즘을 사용하였다. 또한, 각 클러스터에 대해 GO 주석을 OmicsBox v3.4.5를 사용하여 수행하였다. GO 풍부도 분석의 유의성을 평가하기 위해 우리는 Fisher의 정확성 검정을 사용하였으며, p < 0.01에 대한 임계값을 설정했다(Altschul et al. 1990; Al-Shahrour et al. 2004; Götz et al. 2008). 각 클러스터에 대해 상위 세 가지 GO term을 카테고리별로 표시하였다. GO term이 한 클러스터에서 상위 세 가지 GO term으로 식별되지 않더라도 다른 클러스터에서 나타나는 경우 포함하였다. 또한, 우리는 가중 상관 네트워크 분석(Weighted gene co-expression network analysis, WGCNA)을 사용하여 공동 발현 네트워크 분석을 수행하였다(Langfelder and Horvath 2008). 최적의 소프트 power(β) 값을 선택한 R 패키지를 사용했으며, SFT.R.sq ≥ 0.8 및 minModuleSize 30을 설정했다. 네트워크 시각화는 Cytoscape v3.10.3을 사용하여 진행하였다(Shannon et al. 2003).
결과 및 고찰
사과 및 배에서의 MADS-box 유전자군 재동정 및 특징 분석
다양한 유전자군 서열이 실제 유전체 서열내에 존재하지만 유전자 주석정보에 누락되어 있어, 재동정을 통한 유전자 정보 업데이트는 후속 기능연구에 필수적이다(Ouzounis and Karp 2002). 본 연구에서는 선행 연구에서 동정되지 않은 MADS-box 유전자들을 찾아 분석의 정확도를 높이기 위해 TGFam-Finder를 통하여 새롭게 유전자들을 동정하였다(Table 1). 그 결과, M. domestica에서는 이전에 동정된 유전자 101개와 새로 동정된 유전자 47개가 확인되었고, P. pyrifolia에서는 이전에 동정된 유전자 71개와 새로 동정된 유전자 57개가 발견되었다. 반면, A. thaliana에서는 이전에 동정된 유전자 109개와 새로 동정된 유전자 3개가, O. japonica에서는 이전에 동정된 유전자 76개와 새로 동정된 유전자 6개가 확인되었다. A. thaliana와 O. japonica는 모델 식물로서 비교 연구로 활용하기 위해 함께 분석이 진행되었다.
Table 1.
Numbers of Re-annotated MADS-box genes in four plant species
| Species | Previously annotated genes | Newly annotated genes | Total |
| Malus domestica | 101 | 47 | 148 |
| Pyrus pyrifolia | 71 | 57 | 128 |
| Arabidopsis thaliana | 109 | 3 | 112 |
| Oryza japonica | 76 | 6 | 82 |
| Total | 357 | 113 | 470 |
총 4종에서 이전에 동정된 유전자 357개와 새로 동정된 유전자 113개가 확인되었다. 선행 연구가 많이 진행된 A. thaliana와 O. japonica의 경우 새로 동정된 유전자의 비율이 각각 2.7%와 7.3%로 매우 적었지만, M. domestica와 P. pyrifolia에서는 각각 약 31.8%와 44.5%의 새로 동정된 유전자가 확인되어 선행 연구들과 비교하여 유의미한 결과를 얻을 수 있었다. MADS-box 유전자의 type별 새로 동정된 유전자와 이전에 동정된 유전자 개수를 확인하였다(Fig. 1A). MADS-box 유전자의 type은 재동정된 유전자들을 선행연구에 따라 분류하였다(Par̆enicová et al. 2003). MIKC-type의 경우 이전에 동정된 유전자는 146개, 새로 동정된 유전자는 44개였고, M-type의 경우 이전에 동정된 유전자는 211개, 새로 동정된 유전자는 69개였다. MIKC-type 유전자는 꽃의 형성, 발달, 그리고 구조의 패턴 형성 등에 관여하기 때문에, 피자식물군에 속하는 식물들에서는 일반적으로 MIKC-type 유전자가 M-type 유전자보다 더 많이 나타나지만, 본 연구에서는 MIKC-type 유전자가 M-type 유전자보다 적게 나타났다.
각 종에서 MADS-box 유전자의 특징을 분석하기 위해 4종의 MIKC-type과 M-type 유전자의 비율을 확인하였다(Fig. 1B). MIKC-type 유전자의 비율은 P. pyrifolia(43.8%)에서 가장 높았고, 다음으로 O. japonica(42.7%), A. thaliana (38.4%), M. domestica(37.8%) 순으로 나타났다. 4종의 MIKC-type과 M-type 유전자의 개수는 각각 P. pyrifolia(56, 72), O. japonica(35, 47), A. thaliana(43, 69), M. domestica(56, 92)로 집계되었다.
각 type별 MADS-box 유전자에 보존된 서열 구성을 분석하기 위해 MADS-box 도메인에 대한 서열 분석을 진행하였다(Fig. 1C). 분석 결과, α-helix 1개와 β-sheet 2개가 나타났으며, 특히 α-helix는 β-sheet보다 더 높은 보존율을 보였다. MADS-domain을 2차 구조에 따라 7개 부분으로 나누어 각 영역의 consensus 점수를 계산한 결과, MIKC-type이 M-type에 비해 전반적으로 높은 보존 정도를 나타냈다.

Fig. 1.
Characteristics of MADS-box genes in four plant species. (A) The bar graph shows the number of MADS-box genes of each type in four plant species, with domain repertoires represented by symbols on the left. Each bar indicates the MADS-box type, with the bar color reflecting the annotation results. (B) The upper and lower bar charts illustrate the distributions and quantities of MADS-box genes across the four species, with the upper chart showing proportions and the lower chart displaying quantities; red represents the MIKC type and blue represents the M type. (C) The conserved amino acid sequence and secondary structure of the MADS-box domain are presented, with the x-axis indicating the amino acid residue positions and the y-axis showing the corresponding relative abundance levels. Predicted regions of α-helix and β-sheets are indicated by arrows and colored boxes, while the middle amino acid sequence is representative for each species. The bar graph illustrates consensus scores for the secondary structure segments. (D) The heatmap displays the distributions of the top five gene ontology (GO) terms across MADS-box subfamilies, with GO categories on the left and terms on the right. The scale bar below indicates the percentage of each GO term.
재동정된 MADS-box 유전자의 기능을 추론하기 위해 Gene Ontology(GO) 분석을 수행하였다(Fig. 1D). 각 subfamily에 속한 MADS-box 유전자는 ‘biological process’, ‘molecular function’, ‘cellular component’로 분류되었으며, 각 범주에서 우세한 GO는 유사한 경향을 보였다. 주요 GO는 ‘positive regulation of transcription by RNA polymerase II’, ‘protein dimerization activity’, ‘nucleus’였고, molecular function에서 ‘DNA-binding transcription factor activity, RNA polymerase II-specific’도 높은 비율을 나타냈다. 이 결과는 업데이트된 MADS-box 유전자들이 전사인자로서 단백질 이합체를 형성하며, RNA polymerase II를 통해 유전자 발현을 촉진하는 핵심적인 역할을 한다는 것을 시사한다. 이는 이들 유전자가 식물의 발달, 특히 꽃 형성과 관련된 복잡한 유전자 조절 네트워크에서 중요한 역할을 할 가능성을 나타낸다.
마지막으로, MADS-domain 모티프 분석을 통해 각 type별로 MADS-box 유전자가 어떤 모티프로 구성되어 있는지를 조사하였다(Fig. 2). 모티프 분석 시, 도메인 전후 10개의 아미노산을 포함하여 MADS-domain의 보다 확장된 모티프를 얻었다. M-type 유전자는 motif 3-motif 1-motif 2의 보존된 모티프 구성을 가지지만 아미노산의 보존 정도는 낮았다(Fig. 2A). MIKC-type 유전자는 motif 3-motif 1-motif 2-motif 4의 보존된 모티프 구성을 가지고 있었으며 이 패턴은 MIKC-type 유전자들 사이에서 매우 높은 아미노산 보존성을 보였다(Fig. 2B). M-type의 모티프 구성은 MIKC-type의 motif 3-motif 1-motif 2-motif 4 구조에서 마지막 motif 4가 없어진 형태로 나타났으며, 이는 진화 과정에서 MIKC-type으로부터 변화하여 M-type으로 분화했을 가능성을 시사한다. MIKC-type 유전자는 I-domain이라는 특정 구조 영역을 가지고 있는 반면, M-type 유전자에서는 이 부분이 보존되어 있지 않았다. 결론적으로, 모티프 패턴과 도메인 구조의 차이를 통해 볼 때, M-type MADS-box 유전자는 MIKC-type에서 도메인 변형을 거쳐 진화적으로 파생되었음을 시사한다.
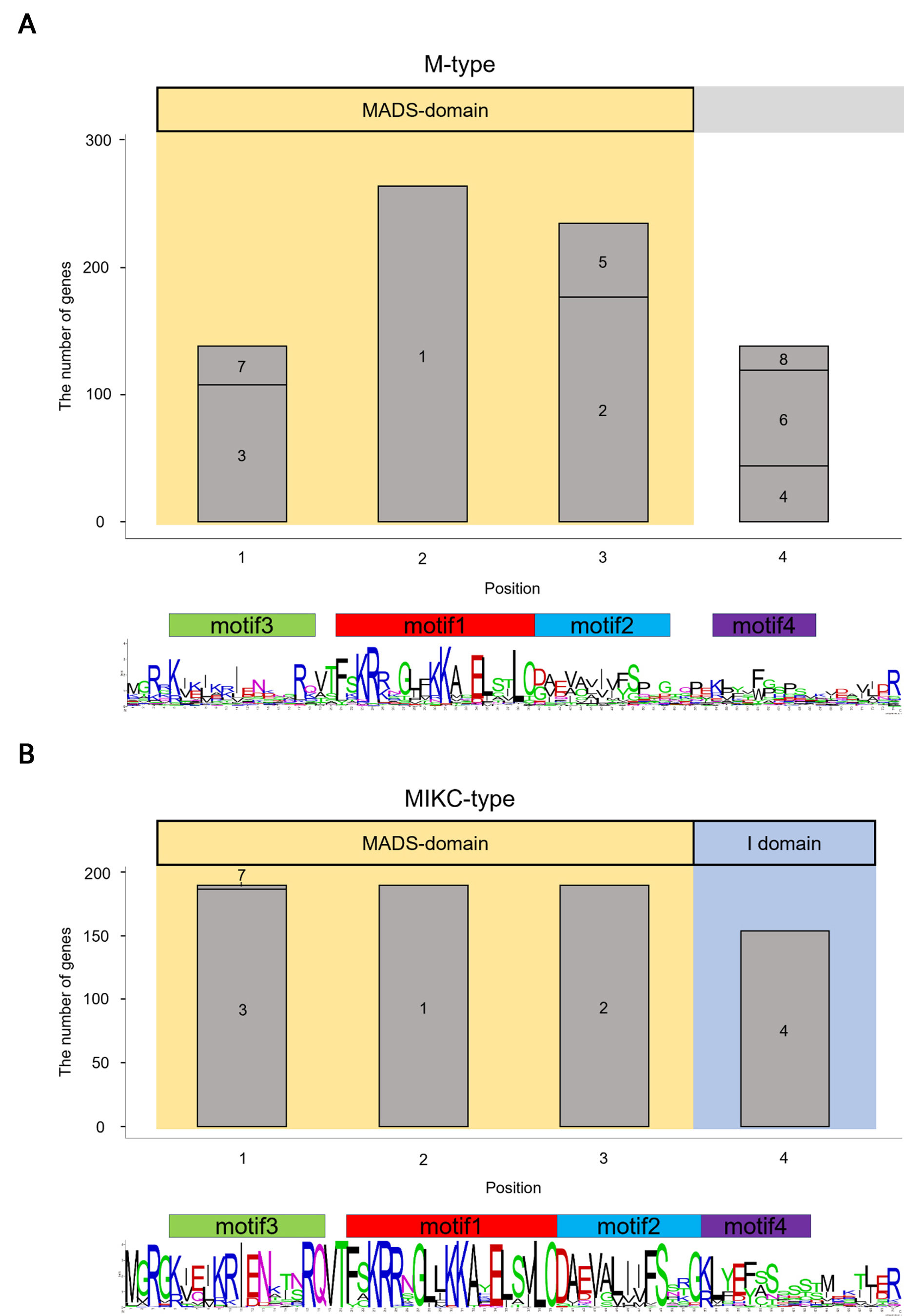
Fig. 2.
Motif compositions of MADS-box genes in four plants species. (A) The motif compositions of M-type MADS-box genes are shown. The x-axis represents the positions of the amino acid sequence, while the y-axis indicates the number of MADS-box genes containing each corresponding motif. The numbers inside each box are motif numbers. The region highlighted in the yellow box indicates the positions of the MADS domain within the sequence. (B) The motif compositions of MIKC-type MADS-box genes are shown. The position belonging the I domain is shown in the blue box.
MADS-box 유전자의 계통수와 CAFE 분석
MADS-box 유전자의 진화적 관계를 조사하기 위해 계통수 분석을 수행하였다(Fig. 3A). MADS-box 유전자의 subfamily는 A. thaliana와 M. domestica의 선행 연구를 기준으로 정립하였으며(Kumar et al. 2016), 분석에 사용된 종은 M. domestica, P. pyrifolia, A. thaliana, O. japonica이다. 이 중 A. thaliana와 O. japonica는 비교군으로 사용되었다.
총 470개의 MADS-box 유전자를 대상으로 계통수를 작성한 결과, 전체 subfamily 중에서 Mα가 21.1%로 가장 큰 비율을 차지하였고, 다음으로 Mγ(13.4%)와 Mβ(9.79%)가 뒤를 이었다. MIKC-type 유전자의 비율은 SOC1(8.51%), MIKC*(7.66%), AP1(6.81%) 순으로 나타났으며, FLC 유전자는 A. thaliana에만 존재하여 1.28%로 가장 낮은 비율을 보였다.
각 종에서 어떤 subfamily가 우세한지를 알아보기 위해 4종의 subfamily별 MADS-box 유전자 수를 조사하였다(Fig. 3B). 모든 종에서 Mα에 속하는 유전자의 수가 가장 많았고, 두 번째로 많이 나타나는 subfamily는 종마다 달랐다. 예를 들어, P. pyrifolia의 경우 SOC1이 두 번째로 많은 subfamily였지만, 다른 종에서는 SOC1의 유전자 수가 상대적으로 적었다. 이러한 차이는 MADS-box 유전자들이 계통 특이적으로 복제되었음을 시사하며, 특히 P. pyrifolia의 SOC1 유전자가 다른 종의 SOC1 유전자와는 다른 종 특이적인 기능을 가질 가능성을 나타낸다. 이와 같은 계통 특이적인 복제는 MADS-box 유전자 레퍼토리의 다양성 생성에 기여할 수 있다.
계통발생학적 증거에 기반하여 유전자 복제 과정에서 각 subfamily가 어떻게 확장되었는지를 조사하기 위해 CAFE(Computational Analysis of gene Family Evolution) 분석을 진행하였다(Fig. 3C). O. japonica가 분화된 이후, M. domestica, P. pyrifolia, A. thaliana의 계통에서 SOC1, AP1, SVP, SEP 등의 subfamily가 확장되었으며, M. domestica는 SVP, AP1, Mγ, AGL17, MIKC*가 확장되었고, P. pyrifolia는 SOC1, AGL6, AP3/PI, TT16이 확장되었다. 분화 전후를 포함하여 M. domestica에서는 AP1과 SVP가 가장 크게 확장되었고, P. pyrifolia에서는 SOC1이 가장 크게 확장되었다. 이는 확장 정도와 시기를 통해 각 종에서 어떤 subfamily가 종 특이적인 형질을 발현할지 예측할 수 있게 한다. 앞서 subfamily별 유전자 개수와 함께 CAFE 분석 결과에서도 P. pyrifolia에서 SOC1의 확장이 가장 두드러지므로, SOC1이 해당 종에서 종 특이적인 기능을 할 확률이 높다고 생각된다. 따라서 이후 발현 분석에서는 P. pyrifolia의 SOC1을 중점적으로 살펴보기로 하였다.
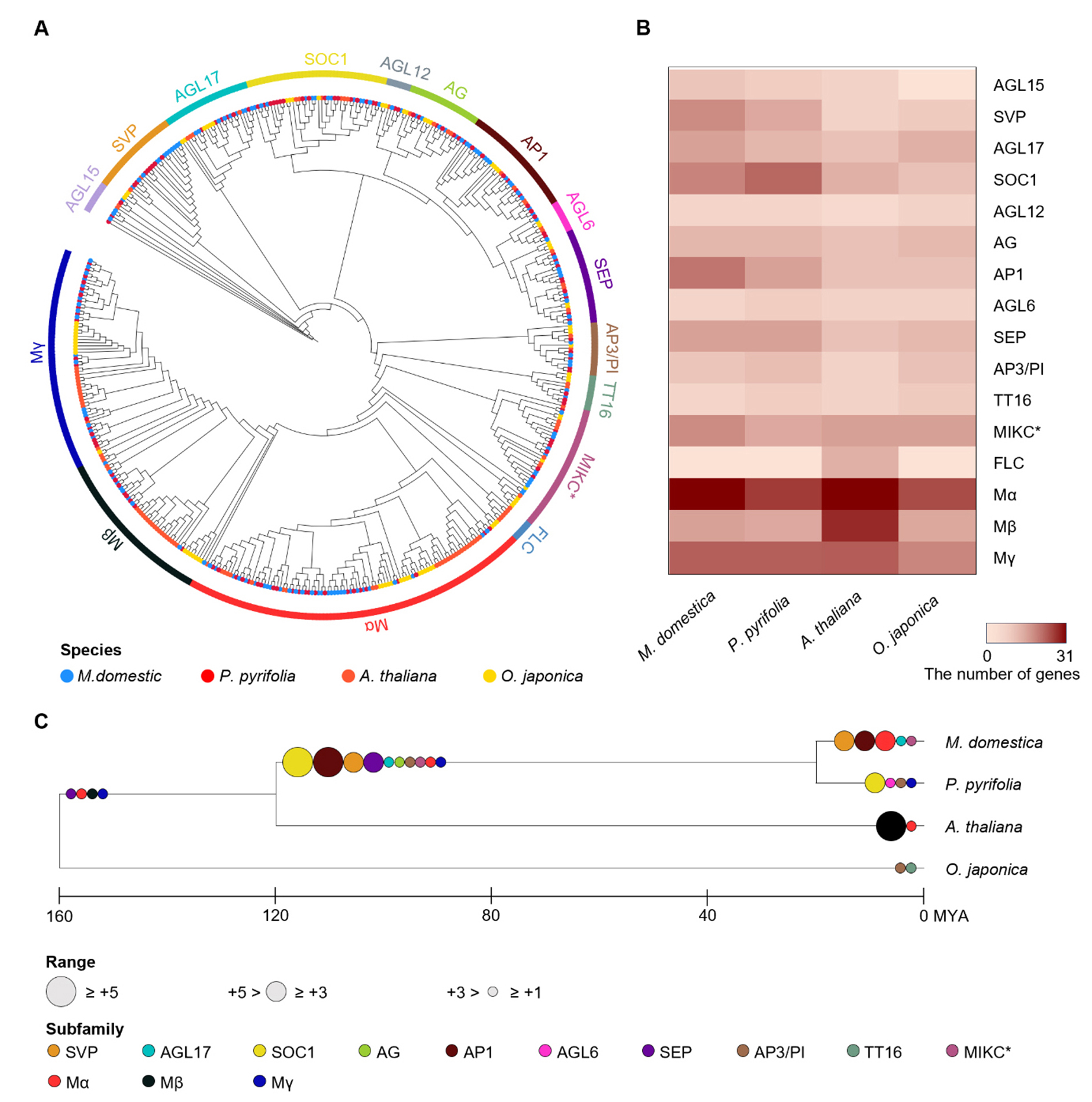
Fig. 3.
Phylogenetic relationships and expansion of MADS-box genes. (A) Phylogenetic tree of MADS-box genes in four species. The dots within the circle indicate the species of the genes, while the outer circle represents the subfamilies. (B) The number of MADS-box genes in each subfamily by species is shown in a heatmap. The scale bar below the species indicates the number of genes. (C) Copy number expansion of MADS-box genes by subfamily across four plant species. The colors and sizes of the circles on the branches reflect the subfamilies and the degree of copy number expansion, respectively.
P. pyrifolia에서 저온 스트레스에 따른 MADS-box 유전자의 발현 및 기능 분석
개화기에 저온은 생식 기관에 손상을 주고 생리장애를 유발하여 생산량과 품질을 크게 저하시킨다(Kwon et al. 2022). 따라서 본 연구에서는 저온 스트레스 조건에서 P. pyrifolia의 MADS-box 유전자의 잠재적 역할을 이해하기 위해 유전자 발현 분석을 수행하였다. 저온 스트레스를 받는 세 시점(15일, 30일, 45일)에서의 발현 프로파일을 조사하였고, MADS-box 유전자의 발현 패턴을 차등 발현 유전자(Differentially expressed genes, DEGs)와 비교하기 위해 전체 P. pyrifolia의 MADS-box 유전자와 3,487개의 DEGs를 포함한 발현 클러스터링 분석을 실시하였다. 이 유전자들은 각각 유사한 발현 패턴을 보이는 네 개의 클러스터로 그룹화되었다(Fig. 4A). 이때 C2와 C4 클러스터는 저온 처리 후 15일에서 45일로 가면서 발현량이 증가하거나 감소하는 경향을 보였다. 이는 해당 클러스터들이 저온 스트레스에 따라 발현이 조절됨을 나타낸다.
P. pyrifolia에서 128개의 MADS-box 유전자 중 68개가 클러스터링 되었으며, 각 클러스터에서 C1 14개(2.60%), C2 16개(3.19%), C3 17개(4.13%), C4 21개(5.04%)로 분포하였다(Fig. 4B). 각 클러스터의 subfamily별 유전자의 분포를 조사한결과, C4에서 SOC1 유전자가 8개로 가장 두드러지게 나타났다. 이는 SOC1이 일정한 발현 패턴을 보이며 저온 스트레스에 반응하는 형질을 나타낼 가능성을 암시한다. 따라서 이후 분석은 SOC1 중심으로 수행하기로 하였다.
P. pyrifolia에서 MADS-box 유전자의 저온 스트레스와 관련된 기능을 추정하기 위해 Gene Ontology(GO) enrichment 분석을 수행하였다(Fig. 4C). 그 결과, 저온 조건 하에서의 발달 및 생식 관련 GO term이 확인되었다. 구체적으로, 발달 관련 GO term으로는 Developmental process(GO:0032502)와 Multicellular organism development(GO:0007275)가 포함되었다. 기존 연구에 따르면, 저온 스트레스는 식물의 성장과 발달을 억제하는데 이는 식물이 성장 대신 생존에 에너지를 우선적으로 사용하기 때문이다. 더욱이, 발아, 잎의 확장, 뿌리의 성장 등의 생리적 과정등이 저해되어 식물 발달을 억제할 수 있다(Zinn et al. 2010). 예를 들어, OsMADS57은 OsTB1과 함께 Dwarf14의 발현을 억제하고 온도 의존적으로 OsWRKY94의 발현을 활성화하여 기관 형성을 방어 반응으로 전환하고 저온 저항성을 증진시키는 것으로 알려졌다(Arora et al. 2007; Chen et al. 2018).
한편, 생식 관련 GO term으로는 Developmental process involved in reproduction(GO:0003006), Reproductive process (GO:0022414), Reproductive system development(GO:0061458), Reproductive structure development(GO:0048608)가 포함되었다. 적절한 개화 시기는 식물의 생식적 성공에 중요한 요소로, 저온 조건에서 꽃이 피면 꽃과 생식 기관이 제대로 발달하지 못해 수분 작용 및 수정 과정에 문제가 생길 수 있다(Seo et al. 2009). 따라서 식물은 저온 환경에서 개화를 지연한다. SOC1은 저온 스트레스 신호 전달에 관여하는 CBF 유전자의 프로모터에 결합하여 전사를 억제하여, 식물의 저온 내성을 높이는 COR 유전자의 발현을 줄인다(Seo et al. 2009). 또한, 식물의 녹화 및 내한성에 관여하는 GNC와 GNL 전사인자의 발현을 억제하여 이들이 유도하는 CBF 및 COR15의 발현도 간접적으로 낮춘다(Richter et al. 2013). 결과적으로, SOC1의 발현이 감소하면 이러한 억제 효과가 해제되어 개화 지연을 비롯한 식물의 저온 반응이 강화된다(Seo et al. 2009; Richter et al. 2013). 이러한 결과는 P. pyrifolia SOC1의 저온 저항성에 대한 기능을 유추할 수 있게 한다.
추가적으로, 차등 발현된 MADS-box 유전자와 DEGs 간의 공동 발현 관계 네트워크 분석을 통해 SOC1에 속하는 PPYR_MADS_102와 PPYR_MADS_56이 각각 56개와 25개의 DEGs와 공동 발현하는 것으로 나타났다(Fig. 4D). PPYR_MADS_102는 저온 내성을 향상시키는 데 관여한다고 알려진 NAM(Li et al. 2016), AUX/IAA(Lu et al. 2024), Myb(Liu et al. 2023) 및 Transferase(Yang et al. 2016) 도메인을 포함하며 이들과의 공동 발현은 PPYR_MADS_102에 저온 저항성에 관한 기능이 있을 것을 시사한다.
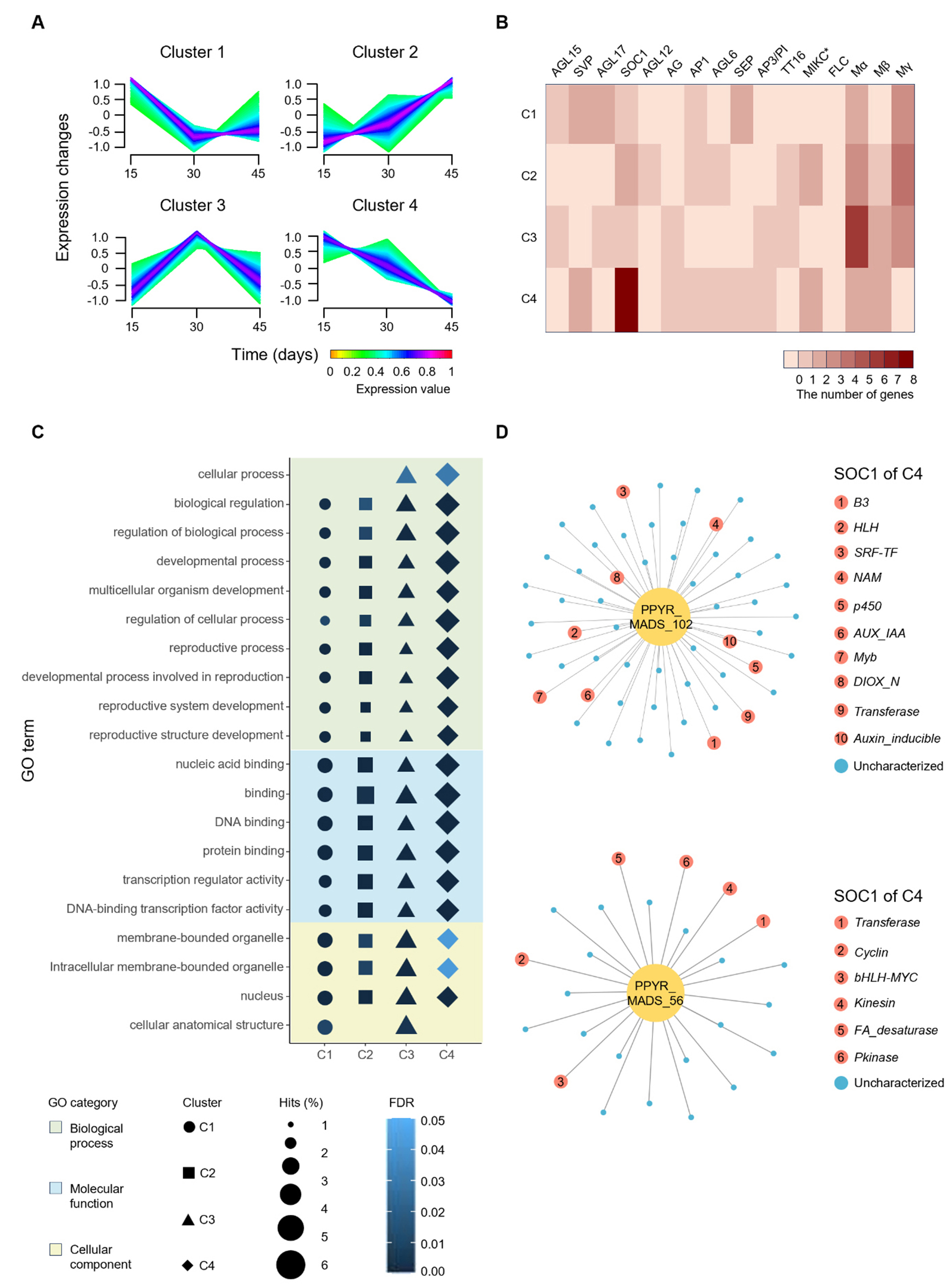
Fig. 4.
Expression clustering and co-expression networks of MADS-box genes in P. pyrifolia under cold stress. (A) Expression patterns of DEGs and MADS-box genes during cold stress. (B) The heatmap displays the number of MADS-box genes for each subfamily in each expression cluster. (C) Dot plot of the top three GO terms in each cluster and category. The background colors represent the three GO categories, and the shape and size of the symbols correspondingly indicate the different clusters and frequency of GO terms. The dot colors illustrate the FDR (false discovery rate). (D) Co-expression networks of MADS-box genes under cold stress are shown. Red dots indicate domains that are co-expressed with the central gene, while light blue dots indicate uncharacterized genes.
마찬가지로, PPYR_MADS_56은 저온 저항성을 높이는 데 관여하는 Transferase, bHLH-MYC(Xu et al. 2014), FA_desaturase(Khodakovskaya et al. 2006) 및 protein kinase(Wang et al. 2025)도메인을 가지는 DEGs와의 공동 발현을 하였다. 결론적으로, 위의 결과들로 볼 때 SOC1은 P. pyrifolia의 저온 스트레스 반응에서 중요한 역할을 담당할 가능성이 높다.
본 연구에서는 사과와 배의 MADS-box 유전자를 재동정하고 그 특성을 분석하였다. 계통수와 CAFE 분석 결과, P. pyrifolia의 SOC1이 특히 두드러진 모습을 보였다. 그러므로 이후 전사체 분석에서 SOC1을 활용하여 연구를 진행하였으며, 그 결과 P. pyrifolia의 SOC1에 속하는 MADS-box 유전자들이 저온 스트레스에 저항하는 기능을 할 가능성이 높다는 것을 시사하였다. 발굴된 유전자는 배의 저온 저항성 관련 유전자 후보로 활용될 수 있으며, 우리의 연구 결과는 향후 사과와 배의 유전체 분석 및 육종 연구에 크게 기여할 것으로 기대된다.
