

Introduction
Materials and Methods
Sample and Preparation
Cell Culture
Cell Viability Assay
Determination of NO Production
Western Blotting
Real-time Polymerase Chain Reaction (PCR)
Statistical Analyses
Results and Discussion
Cell Viability by B. tinctoria Root Treatment
Inhibitory Effect of B. tinctoria Root Extract Treatment on NO Production
Inhibitory Effect of iNOS and COX-2 Protein Expression by B. tinctoria Root Treatment
Inhibitory Effects of B. tinctoria Root Treatment on Cytokines with Real-time PCR
Conclusions
Introduction
Wild indigo (Baptisia tinctoria) dye is an important medicinal raw material containing many biologically active substances and a wide spectrum of pharmacological activity. The genus Baptisia has about 20 species; thus, identifying raw materials of the Baptisia genus for producing medicines is urgently needed. Furthermore, this plant has alternative effects for disease-fighting (Lyle, 1897; Hoffmann, 1996; Duke, 2002; Bone, 2003), such as febrifuge, immune stimulant (Hutchens, 1991), antiseptic (Chevallier, 2001), anti-parasitic, hepatic tonic (Finley-Ellingwood, 1919), choleretic, cholagogue, laxative, lymphatic, and emmenagogue activities. Additionally, pharmacological properties have been observed (Macht and Black, 2007). For example, derivatives of glycoproteins from B. tinctoria show immunomodulatory activity that stimulate lymphocyte DNA synthesis (Beuscher et al., 1989).
The Anti-inflammatory response is a complex response to skin tissue or human body injury and involves T-cell and numerous mediators (Cho and An, 2008). Our body is constantly exposed to bacteria, viruses, fungi, parasites, and other infectious and toxic agents. Neutrophils and tissue macrophages, such as Raw 264.7 cells, attack and destroy invading bacteria, viruses, and other harmful agents (Fujiwara and Kobayashi, 2005). There are three similar forms in nitric oxide synthase (NOS) in mammalian cells: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS). Although eNOS and nNOS are always produced in the body, iNOS is induced by inflammatory factors (Nathan, 1992). Notably, iNOS results in the imbalance of NO concentrations, resulting in NO activating guanyl cyclase and cell toxicity (Lee et al., 2007). Cyclooxygenase-2 (COX-2) and prostaglandin E synthase 2 (PTGES2) are both involved in the production of prostaglandin E2 (PGE2), which has both pro- and anti-inflammatory effects. Furthermore, physiological stress and anti-inflammatory responses arise due to NO production by iNOS in the presence of pro-inflammatory mediators (Yun et al., 2007). NO production is involved in many biological processes, such as the immune system, cytotoxic neurotransmitter system, and vascular relaxation (Kim et al., 2004a). However, excess NO formation results in inflammation, gene mutations, and organ or nerve damage (Moncada et al., 1991; McCartney-Francis et al., 1993).
When anti-inflammation is activated in the body, inflammatory Raw 264.7 cells stimulated with lipopolysaccharide (LPS) and macrophage cells, secretes inflammatory mediator proteins, pro-inflammatory cytokines, and chemokines (Lee and Cho, 2021). The synthesis of pro-inflammatory cytokines, such as interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and IL-6, is controlled by endogenous antagonists for inflammation intermediation. PTGES2 is produced by the COX-2 enzyme, which promotes inflammation, fever, and pain. COX-1 is the only enzyme that creates PGE2, a substance that activates platelets and protects the stomach and intestinal walls (Lee et al., 2020). When inflammation occurs, macrophages, which migrate to the site of inflammation, recognize LPS. Toll-like receptors, which are essential LPS receptors for signal transduction, increases the activity of cytokine genes, such as TNF-α, IL-6, and IL-1β (Kim et al., 2004b).
From clinical outcome studies, no precise efficacy of B. tinctoria roots can be drawn. Therefore, additional research requires using B. tinctoria in monotherapy and determining the inhibitory effect involved in inflammation to determine efficacy.
Materials and Methods
Sample and Preparation
B. tinctoria root cultivars were grown in Fujian Province, China, and the geographical coordinates are 27°32'36"– 27°55'15" north latitude; 117°24'12"–118°02'50" east longitude. Plants were obtained from the Joryherb Biotechnology Co., Ltd. (Shaanxi, Xi’an, China). The B. tinctoria root used as a material was dried in a 45ºC dry oven (Jeiotech, Daegu, South Korea), then using high-speed grinder (RT-08, Rong Tsong Precision Technology, Taichung, Taiwan) at 25,000 rpm. It was pulverized and filtered through a 40-mesh sieve to form a powder. The roots were ground into powder and stored at 4°C, and the extract was dissolved in distilled water and 50% ethanol. A solution was made and incubated at −80ºC for 4 d in a freeze dryer (FDS8518; Ilshin Bio Base Co. Ltd. Dongducheon, Korea). The dried powder was made soluble and divided into different concentrations from 5 to 100 µg·mL-1. Also, extracts of solid dissolved in water and 50% ethanol solution (1:100) and added to B. tinctoria root powder at room temperature in a shaking incubator for 24 h. The extract was filtered with filter paper (No. 1, Whatman Inc., Piscataway, New Jersey, USA) and concentrated using a rotary vacuum evaporator to remove the organic solvent. After freeze-drying, the concentrated extract was dissolved in Dulbecco's modified Eagle’s medium (DMEM) (GE Healthcare Life Sciences, Chicago, IL, UT, USA) prepared from 5 to 100 µg·mL-1.
Cell Culture
Raw 264.7 cells are derived from mice and bought from the American Type Culture Collection (TIB-71, Manassas, VA, USA). Each cell, during the experiments, was used and cultured in DMEM, prepared with 10% fetal bovine serum (FBS) (GE Healthcare Life Sciences, Chicago, IL, UT, USA) and supplemented with 1% penicillin-streptomycin (100 U/mL) (GE Healthcare Life Sciences), plated on cell culture dishes and in atmosphere 5% CO2 at 37°C incubated moisturizing incubator (311, Thermo Fisher Scientific, Rockford, IL, USA). An inverted microscope, Mlchrome 20 (Nikon Inc., Tokyo, Japan), was used for supervision cells and all culture cells in the assays were sub-cultured in a 100 mm × 20 mm culture dish when grown to a density of about 80%, and only cells not exceeding 20 passages were used. Pretreatment of the sample solution in Raw 264.7 cells was performed for 10 min in the cell culture dish washed with phosphate-buffered saline (PBS) (Thermo Fisher Scientific, Rockford, IL, USA). Finally, 1 µg·mL-1 of LPS-stimulated was treated for 18–20 h.
Cell Viability Assay
Cell viability was measured following the methods of Carmichael et al. (1987). The crude cell was seeded in 48-well plates at a density of 5 × 104 cells/well and cultured for 24 h at 37°C in a 5% CO2 incubator. Then, 50 µL of samples at different concentrations (B. tinctoria 5–50 µg·mL-1) was added and incubated for 18 h at 37°C with 5% CO2. Next, 50 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 µg·mL-1, Sigma Chemical Co., St. Louis, MO, USA) was added and reacted for 4 h. After the medium of each well was removed, 500 µL dimethyl sulfoxide (Junsei Chemical Co. Ltd., Tokyo, Japan) was added and room temperature for 30 min to respond. The ELISA reader (SPECTROstar Nano, BMG LabTech, Ortenberg, Germany) was used to measure the absorbance at 540 nm.
Determination of NO Production
The cells were seeded in 96-well plates at a density of 1 × 105 cells/well and incubated in a culture medium for 24 h at 37°C in a 5% CO2 incubator. Then, the cells were stimulated with 1 µg·mL-1 of LPS (20 µL), followed by the addition of 20 µL B. tinctoria (5–50 µg·mL-1) and incubated at 37°C in a 5% CO2. After 18 h, 0.1 mL of supernatant was reacted using the Griess reagent system (Promega, Madison, WI, USA), and the measure of absorbance was read at 540 nm to determine the NO production (Ryu et al., 2003; Cho and An, 2008).
Western Blotting
To investigate the expression of iNOS and COX-2 proteins, the Raw 264.7 cells were seeded in 6-well plates (5 × 105 cells/well), taken after by hatching were cultured for 24 h in culture DMEM at 37°C in a 5% CO2 incubated. The medium was withdrawn at the end of stabilization, and scrubbed with PBS all wells and stimulated with 1 µg·mL-1 of LPS. After 10 min, 10, 30, and 50 µg·mL-1 of B. tinctoria was extracts treated and cultivated for 20 h. After the medium was removed and scrubbed twice with PBS. Protein was eluted with 100 µL lysis buffer, 70 µL mammalian protein extraction reagent was added and centrifuged at 12,000 rpm for 20 min at 4°C. The supernatant was retrieved, and protein content was determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Protein (40 µL) was roading to 10% sodium sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and incubated with 5% skim milk blocking buffer in Tris-buffered saline tween 20 (TBST) for 1 h at room temperature. Next, the membranes were incubated with diluted primary antibodies at 4°C overnight. The primary antibodies were iNOS (1:1,000 in 3% skim milk; #sc-651; Santa Cruz Biotechnology, Dallas, TX, USA), COX-2 (1:1,000 in 3% skim milk; #33345; Signalway Antibody Co. Ltd., Maryland, College Park, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1,000 in 3% skim milk; #GA1R; Thermo Fisher Scientific). The membranes were washed with TBST, and then secondary antibodies were mouse anti-rabbit lgG, heavy and light chain-linked antibody (1:1,000 in 3% skim milk; #sc-2357; Santa Cruz Biotechnology), and goat anti-mouse IgG, (1:1,000 in 3% skim milk; #31430; Thermo Fisher Scientific), and reacted for 1 h. Finally, protein bands were detected using West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific), and images of the blots were captured by ImageJ 1.51j8 (Wayne Rasband, National Institutes of Health, USA) on each band (Cho, 2011).
Real-time Polymerase Chain Reaction (PCR)
RNA expression of B. tinctoria in Raw 264.7 cells stimulated with LPS was used in the experiment by culturing 5 × 106 cells/well in a 100 mm × 20 mm culture dish. Cells were treated with 1 µg·mL-1 of LPS and sample group of B. tinctoria concentrations 10, 30, and 50 µg·mL-1 and re-cultured for 20 h and washed with PBS. Total RNA was extracted using the TRI-Solution RNA extraction kit (BioScience Technology, Daegu, Korea) to compare the mRNA expression of pro-inflammatory factors. Additionally, cDNA was synthesized using a qPCRBIO cDNA synthesis kit (PCR Biosystems, London, UK). The synthesized cDNA was diluted to 1 µL synthesized cDNA and 5 µL using real-time PCR universal qPCR master mix #m3003s (New England BioLabs Inc. Hitchin, UK). Also, by 0.3 µL forward and reverse primer sequence and nuclease-free water until up to 10 µL final volume. Real-time PCR assay of pro-inflammatory factors for each gene was analyzed using a PCRmax Eco 48 real-time PCR system (PCRmax Inc., West Midlands, UK). Real-time PCR conditions and primer sequences are shown in Table 1.
Table 1.
Sequences of primers used for real-time PCR assay
Statistical Analyses
Anti-inflammatory agents (n = 3) were measured, and all assay results were verified by IBM SPSS Statistics Client Documentation 25.0 for Windows (Statistical Package for Social Science, Chicago, IL, USA). Additionally, Duncan's multiple range tests were performed to analyze differences. A p-value of less than 0.01 (p < 0.01) was considered statistically significant.
Results and Discussion
Cell Viability by B. tinctoria Root Treatment
Cell viability is a hypersensitive and exact assay, with color and cell morphology highly correlated with Raw 264.7 cell condition as a measure of toxicity (Keiser et al., 2000). Cell viability assay was performed to check the viability of Raw 264.7 cells treated with B. tinctoria water and ethanol root extract reactions at different concentrations (5, 10, 20, 30, 50, 100 µg·mL-1), and the results are shown in Fig. 1A and 1B. Cell viability were significantly increased cell toxicity at the 100 µg·mL-1 concentration in both the water and ethanol extracts (81.97%–72.45%) as a previous study with sheng-mai yin and amber extracts (Zheng et al., 2021; Tian et al., 2021). For further studies selected concentrations of 5–50 µg·mL-1 were showed non-toxic inhibition. Most of the high cell viability (p < 0.01) in B. tinctoria showed under the 50 µg·mL-1 concentrations of both extracts (>85% cell viability) respectively. cell death due to the cytotoxicity of the extract confirms these results, morphological changes were also observed with a microscope at 24 h after sample treatment (Fig. 1C).
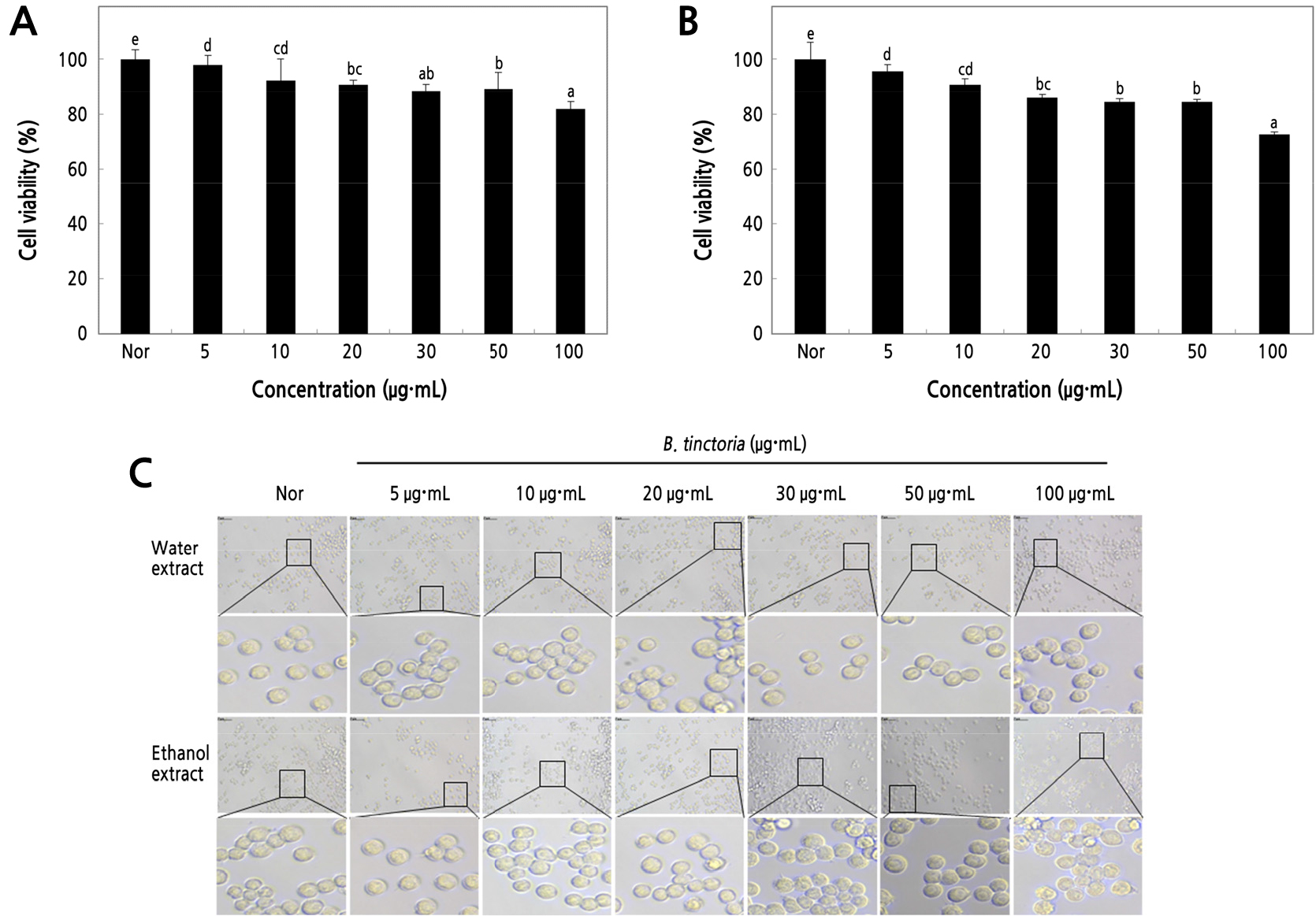
Fig. 1.
MTT assay of water (A) and ethanol extracts (B) from B. tinctoria root. The Raw 264.7 cells (5 × 104 cells/well) were incubated with various concentrations of B. tinctoria from 5 to 100 µg·mL-1 for 18 h. (C) The morphology of cells observed by phase contrast microscopy was photomicrograph modification at 50 µm (magnification, × 400). A negative control group (Nor) was obtained in the absence of B. tinctoria. The result was signified as an average ± standard variation denoted with different letters, which are significantly different at p < 0.01 by Duncan’s range test (n = 3).
Inhibitory Effect of B. tinctoria Root Extract Treatment on NO Production
The macrophage Raw 264.7 cells involved in the inflammatory response, are activated by LPS-stimulator various concentrations for producing pro-inflammatory cytokines such as NO to induce inflammatory responses (Han et al., 2021). Raw 264.7 cells treated with lower concentrations (5, 10, 20, 30, 50 µg·mL-1) of B. tinctoria water and ethanol root extracts and with 1 µg·mL-1 of LPS-stimulated. However, NO expression reduced by 66.82% following exposure to 50 µg·mL-1 (Fig. 2B) more than low from water extract (77.27%) in the same concentrations as shown in Fig. 2A. In a previous study, it was reported that the NO production inhibition effect through iNOS expression inhibition was greater than that of the red beetroot extract by COX-2 inhibition of PGE2 synthesis could be confirmed (Yi et al., 2017).
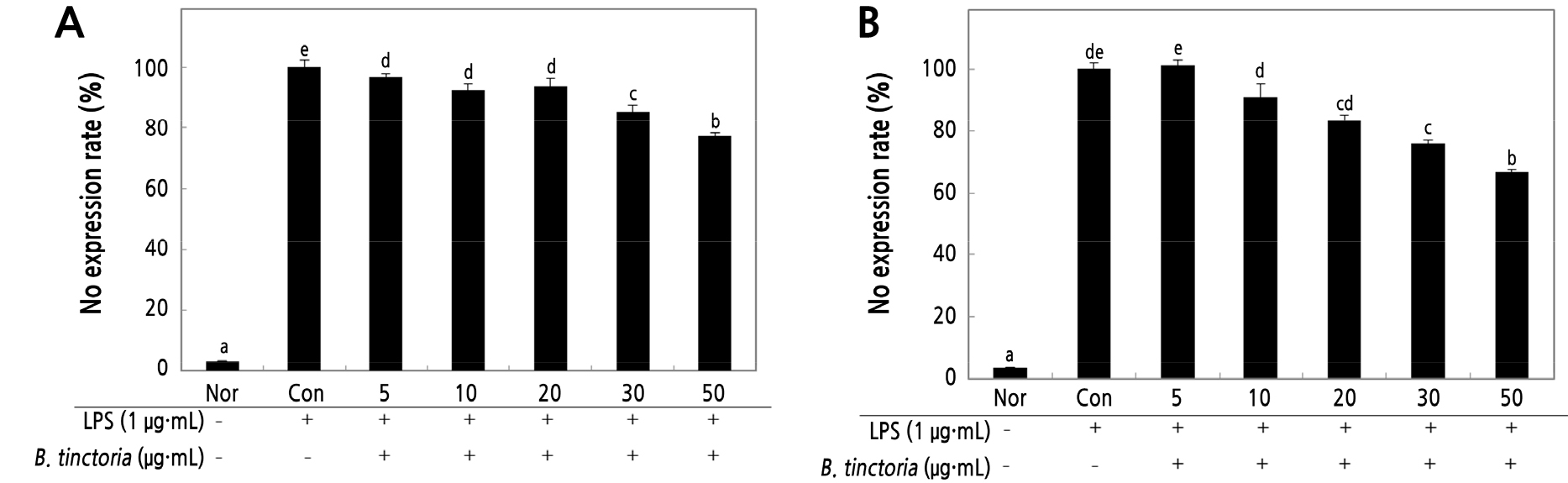
Fig. 2.
Effect of B. tinctoria root on NO inhibitory in Raw 246.7 cells treated with LPS using water (A) and ethanol extracts (B). B. tinctoria extracts ranging from 5 to 50 µg·mL-1 were given to the cells, which were then incubated for 18 h. As a result, LPS was not used to create a normal group (Nor). The negative control group (Con) solely used LPS. Duncan's range test (n = 3) determined that the results were substantially different at p < 0.01 when expressed as an average standard deviation.
Inhibitory Effect of iNOS and COX-2 Protein Expression by B. tinctoria Root Treatment
The inhibitory effects of B. tinctoria root on these pro-inflammatory cytokine mediators such as NO and PGE2 are related to iNOS and COX-2 protein expressions were measured by western blot assay (Fig. 3). In addition, when LPS-stimulated at 1 µg·mL-1 Raw 264.7 cells and treated with B. tinctoria root extracts, there is a link between NO producing and iNOS as well as COX-2 proteins expression significantly inhibited in both of the extracts. Stimulation of macrophage by LPS and were treated at 50 µg·mL-1 concentration of B. tinctoria water extracts, iNOS protein expression decreased by the 6.86% group of positive control (Fig. 3C). However, in ethanol extracts decreased by 47.35% iNOS protein expression at the same concentration.
The upregulation of iNOS and COX-2 expression of protein and COX-2 expression in macrophage cells after LPS-stimulated augment of the production in pro-inflammatory cytokine and chemokine factors, such as TNF-α, IL-1β, fibroblast growth factor, and phosphatidic acid.
Moreover, COX-2 protein induces pro-inflammatory cytokine factors, such as glucocorticoid, IL-4, and IL-13. Consequently, in Raw 264.7 cells, the anti-inflammatory effect can be achieved by decreasing the levels of the COX-2 protein (Surh, 2002; Kim et al., 2004b). B. tinctoria water extracts at 50 µg·mL-1 decreased expression by 54.55% (Fig. 3A).
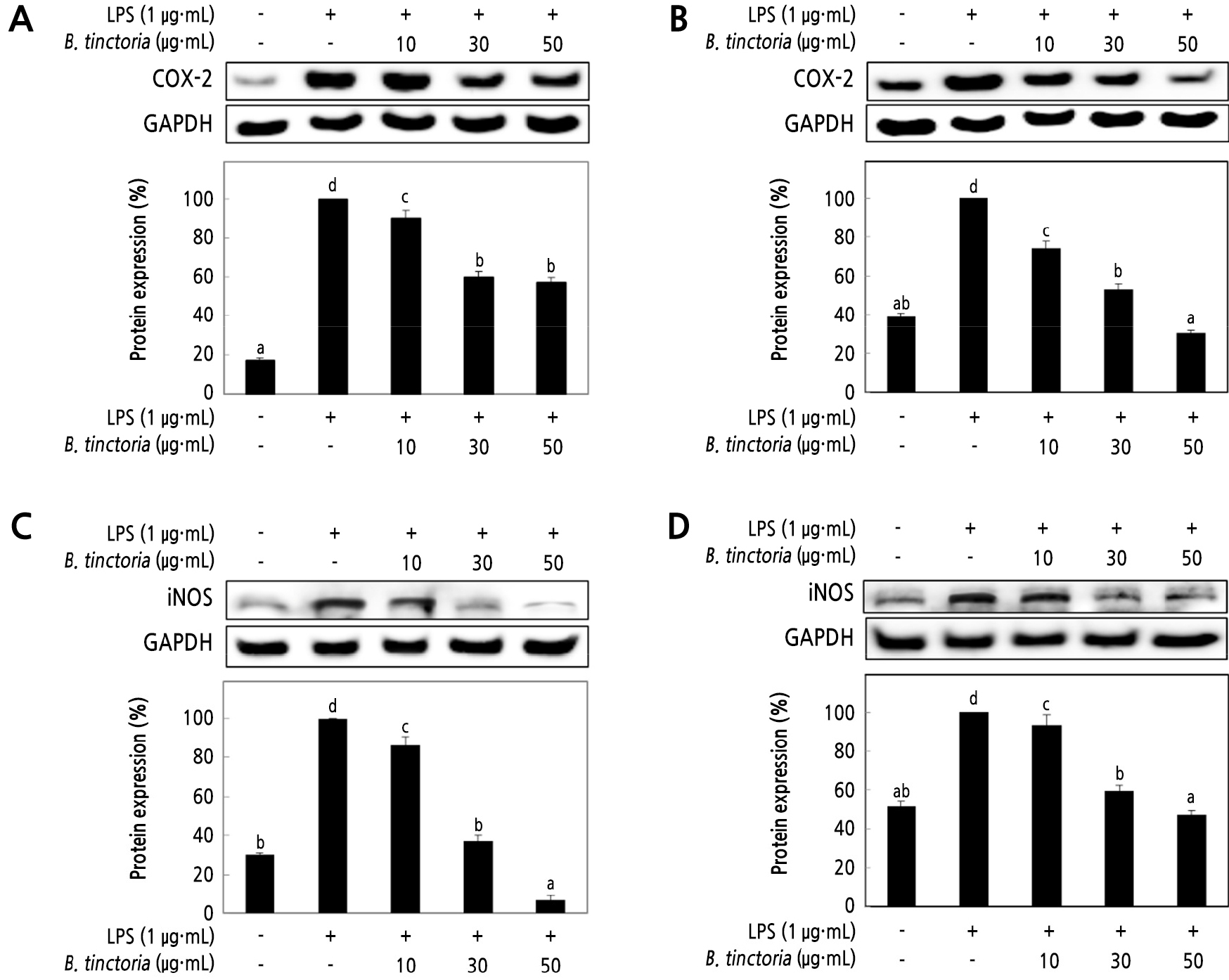
Fig. 3.
The inhibitory effect of water (A) and ethanol extracts (B) from B. tinctoria root on COX-2 protein expression; water extracts (C) and ethanol extracts (D) on the iNOS inhibitory levels in Raw 246.7 cells stimulated with LPS. Raw 264.7 cells (5 × 105 cells/well) were treated with LPS (1 µg·mL-1), and 10, 30, and 50 µg·mL-1 of B. tinctoria was dissolved in DW; the cells were further incubated for 20 h. The normal group was obtained in the absence of LPS. The negative control group treated only LPS. The result was signified as an average ± standard variation denoted with different letters and are significantly different at p < 0.01 by Duncan’s range test (n = 3).
When LPS-stimulated Raw 264.7 cell, toll-like receptor 4 signaling pathway is transferred into the cell and phosphorylation of nuclear factor-kappa β (NF-κβ), an inhibitory protein, and factors related to other proteins, such as iNOS, COX-2, and cytokines (Li and Verma, 2002; Kim et al., 2011). The iNOS and COX-2, which are phosphorylated signaling proteins, are also inhibited. The inhibition of iNOS also showed similar inhibition of NO production. It was also expected to affect the PGE2 or PTGES2 mediated by COX-2. Consequently, a reduction in the level of iNOS protein in macrophages stimulated with LPS is expected to yield an inflammation inhibitory effect. iNOS defends against pathogens and is closely linked with inflammatory disease, circulatory disorder, and cancer.
Inhibitory Effects of B. tinctoria Root Treatment on Cytokines with Real-time PCR
A recent study has been submitted that Pyrrosia lingua extracts suppresses the anti-inflammatory response (Hong et al., 2021). The activity of B. tinctoria as a pro-inflammation inhibitory agent was measured by treatment with 10–50 µg·mL-1 concentration in stimulated Raw 264.7 cells by LPS. Water and ethanol extracts of B. tinctoria showed inhibitory activities on TNF-α, IL-1β, IL-6, MCP-1, and PTGES2 at 50 µg·mL-1 (Fig. 4A–4J). These results confirm that all pro-inflammatory factors are decreased concentration-dependently. Additionally, water and ethanol extract inhibition of MCP-1 and inflammatory cytokines, TNF-α, IL-1β, and IL-6, by both extracts of B. tinctoria are useful in the inflammatory response by macrophage-mediated monocyte aggregation by MCP-1. It was confirmed that PTGES2 was involved in the expression of PGE2.
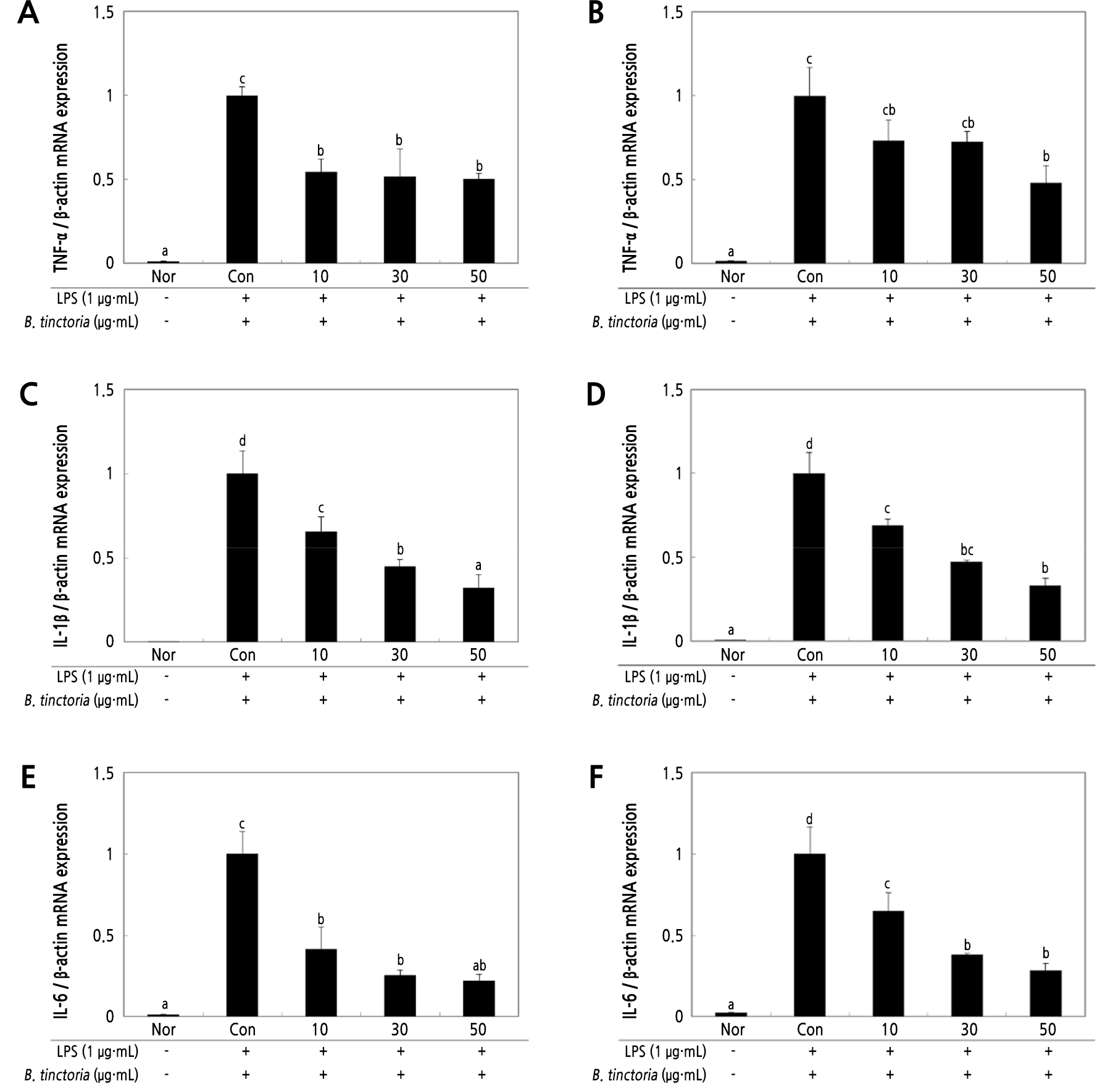
Fig. 4.
The inhibitory effect on the expression of pro-inflammatory cytokines in stimulated Raw 264.7 cells with LPS (1 µg·mL-1). Raw 264.7 cells (5 × 106 cells/well) treated with B. tinctoria water and ethanol extracts at 10–50 µg·mL-1 concentrations were added, and the cells were further incubated for 20 h. The mRNA expression of the pro-inflammatory markers, TNF-α (A, B), IL-1β (C, D), IL-6 (E, F), MCP-1 (G, H), PTGES2 (I, J), was measured by real-time PCR. Relative mRNA gene expression was normalized to β-actin. Normal group (Nor) measurements were obtained in the absence of LPS. The negative control group (Con) treated only LPS. The result was signified as an average ± standard variation denoted with different letters and are significantly different at p < 0.01 by Duncan’s range test (n = 3).
As a result, inhibition patterns of TNF-α, IL-1β, and IL-6 showed similar results with respect to iNOS and COX-2 protein production. Therefore, the anti-inflammatory effect of B. tinctoria was confirmed. The Raw 264.7 cell production and expression of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, MCP-1, and PTGES2 was inhibited and suppressed by B. tinctoria in various concentrations (10, 30, 50 µg·mL-1).
Conclusions
This study evaluated the physiological activities of antioxidants, beauty foods, and functional foods using the extracts of B. tinctoria root. As a result, determined anti-inflammatory effects of B. tinctoria water and 50% ethanol root extracts in Raw 264.7 cells stimulated by LPS were compared to examine the possibility of developing anti-inflammatory materials. The cell viability was measured cell toxicity on Raw 264.7 cells after treatment B. tinctoria root extracts showed positive viability survival rates. The levels of iNOS expression, COX-2 expression, NO generation, and pro-inflammatory cytokine expression were measured to test the anti-inflammatory efficacy. The expression level of iNOS, an enzyme involved in NO biosynthesis in stratified and fermented stratum trees, was associated with an expression-suppression effect of approximately 50% at B. tinctoria root extract concentrations of 10, 30, and 50 µg·mL-1. This is due to measuring the manifestation volume of COX-2 protein, ethanol, and water extract. NO production was greatly inhibited in the B. tinctoria-treated group compared with that in the control group. These results confirmed that B. tinctoria it has potential useful as an anti-inflammatory agent. mRNA expression levels of proinflammatory markers, such as TNF-α, IL-1β, IL-6, MCP-1, and PTGES2, were significantly suppressed.
Furthermore, there was no difference in the mRNA expression of pro-inflammatory cytokines including IL-6, TNF-α, and IL-1β after treatment with low concentration (10 µg·mL-1) of water extract, but their expressions were decreased after treatment with high concentration (50 µg·mL-1) of ethanol extract. In addition, B. tinctoria root extracts can be used in the food industry and are expected to be used in the development of anti-inflammatory substances.
