

Introduction
Materials and Methods
Plant Material Preparation for Eight Accessions
Sequencing of Transcriptomes for Eight Accessions
Quality Trimming of Reads
Reference Sequence Preparation
Repeat Masking on the Reference Sequence
Alignment to the Reference Sequence
SNP Calling and Filtering
Linkage Mapping
Polymorphism Survey and Genetic Diversity
Results
Sequencing of Eight Hot Pepper Accessions and Quality Trimming
Alignment of Each Accession to the Reference Sequence
SNP Discovery and SNP Filtering
Development and Validation of SNP Markers
Discussion
Introduction
Hot pepper (Capsicum annuum) is a crop of major economic importance that is commercially cultivated in China, Korea, the East Indies, and the United States of America, among many other countries (Shao et al., 2008). Worldwide production of hot peppers has been estimated to be 14-15 million tons a year (Weiss, 2002). In fact, hot pepper is the vegetable accounting for the largest planting area in Korea.
In commercial breeding, new traits are commonly intro-duced into elite breeding lines using conventional backcross methods, which involve time consuming efforts to transfer target genes into the genetic background of a recipient parent. Recently marker-assisted backcrossing (MABC), which is more efficient and faster than conventional backcrossing, has been generally accepted as an advanced plant breeding technique (Blum et al., 2002). For successful application of MABC, factors to consider include the number of target genes to be transferred, the genome size of the crop species, and the availability of a highly saturated map. Among those factors, the availability of highly polymorphic markers with known positions in each chromosome is critical (Varshney et al., 2009). Although molecular markers based on restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), and random amplified poly-morphic DNA (RAPD) have been developed and used in practical plant breeding (Imelfort et al., 2009; Jung et al., 2010; Kang et al., 2001; Paran et al., 2004; Yoo et al., 2003), such markers still have limitations for use in MABC due to the difficulty of finding polymorphisms among breeding lines and of high throughput genotyping. Recently, single nucleotide polymorphism (SNP) markers have captured attention because of their potential for high-throughput detection and computerization with automated platforms (Jung et al., 2010; Vignal et al., 2002; Yi et al., 2006).
SNPs are single-base differences in DNA between accessions. In plants, SNPs generally occur in populations once every few hundred base pairs (Metzker, 2005). SNP markers can be developed through several methods. For example, SNPs can be identified by simply comparing a candidate sequence to a reference sequence (Nicolai et al., 2012), by whole genome sequencing (WGS; Goff et al., 2002; The Arabidopsis Genome Initiative, 2000), or by sequence alignment of expressed sequence tags (ESTs) to a reference sequence (Jones, 2009; Kota et al., 2001; Labate and Baldo, 2005). For the crops like hot pepper, in which the genome is huge, ESTs have been adopted as an alternative to WGS and as a substrate for cDNA array-based expression analyses (Kim et al., 2008; Rudd, 2003). ESTs are a few hundred base pairs of sequence derived from randomly selected cDNA clones prepared from specific tissues, and EST sequencing is inexpensive compared to WGS. These characteristics led us to test whether SNP markers could be developed from several pepper accessions using ESTs generated with next generation sequencing (NGS) technology.
NGS is a fast and low cost method for the large-scale generation of reliable and robust transcript sequences and identifying and characterizing genetic polymorphisms in plants (Imelfort et al., 2009; Metzker, 2010). The Illumina Genome Analyzer (IGA) used to be the most widely used platform based on amplified sequencing features generated by bridge PCR (Shendure and Ji, 2008). Since the IGA reads only a short sequences (75-100 base pairs; Flicek and Birney, 2009), SNPs can be identified by either de novo assembly of short sequence reads or alignment to the reference. Several factors can interfere with correct sequence alignment; these include missing calls of overlapping geno-types (Anney et al., 2008), false discovery of polymorphic SNPs (Pettersson et al., 2008), homozygote to heterozygote miscalls (Teo et al., 2007), and allelic dropout (Pompanon et al., 2005).
Here, we describe a process for SNP development from transcriptome sequencing of peppers. The resulting SNP primers clearly distinguished between 27 tested Capsicum cultivars, demonstrating that the SNP markers developed in this study will be useful resources to facilitate MABC in pepper.
Materials and Methods
Plant Material Preparation for Eight Accessions
Jeju (Capsicum annuum), LAM32 (C. annuum), Tean (C. annuum), CM334 (C. annuum), SNU-001 (C. chinense), Yuwolcho (C. annuum), PI201234 (C. annuum) and YCM334 (C. annuum) were grown in a growth chamber with 12 h light at 25°C and 12 h dark at 18°C. Leaf tissues at the same stage from the eight accessions were collected. Total RNA was isolated from leaves of each accession with Trizol extraction buffer (Ambion, Carlsbad, CA, USA) as described in the manufacturer’s protocol and used for sequencing of transcriptomes.
Sequencing of Transcriptomes for Eight Accessions
Jeju, LAM32 and Tean were sequenced via GAIIx sequencing with 116-bp single-end reads at the National Instrumentation Center for Environmental Management (NICEM). CM334 sequencing data of 101-bp paired-end reads were provided by Dr. Choi from the Plant Genomics Laboratory at Seoul National University. SNU-001, Yuwolcho, PI201234 and YCM334 were sequenced with the GAIIx sequencing platform using the 90-bp paired-end read sequencing method at Beijing Genome Institute (BGI).
Quality Trimming of Reads
The sequence data from each accession were trimmed to reduce the quality deterioration in the 3’-end and 5’-end regions, which negatively affects mapping and assembly. Sickle version 1.0 (https://github.com/najoshi/sickle) was used for quality trimming.
Reference Sequence Preparation
The reference sequence, assembled by commercially available CLC Genomics Workbench software, comprised 31,196 contigs from EST sequences that were mainly derived from Korean F1 hybrid line Bukang, for which data are available at the Korea Research Institute of Bioscience & Biotechnology (KRIBB) (Ashrafi et al., 2012; Kim et al., 2008).
Repeat Masking on the Reference Sequence
Repeat masking was performed on the reference sequence using the 727 repeat sequence library set up by the Plant Genomics Laboratory at Seoul National University because of the presence in Capsicum of areas highly abundant in repeat sequence (Yi et al., 2006). The data were collected by the RMBlast program, which is mainly used in NCBI for finding matches between two sequences and attempting to start alignments from these matched places (Johnson et al., 2008). RepeatMasker from the Institute for Systems Biology was used to screen DNA sequences for repeats and areas of low complexity.
Alignment to the Reference Sequence
Sequence alignments to the reference sequence were performed to find SNPs (McPherson, 2009) using the Burrows-Wheeler Transform algorithm from Burrows- Wheeler Aligner version 0.5.9rc1, which requires little memory and so is suitable for reducing the analysis time (Li and Durbin, 2009).
SNP Calling and Filtering
SNP data were collected using SAMtools software and an in-house python script with strict criteria (Li et al., 2009). SNPs from sequencing depth of 25x were filtered in the following order: 1) diallelic SNPs, 2) SNPs found in more than six accessions, 3) SNPs with high PIC value, 4) SNPs for Fluidigm probe design (at least 60 bp from any intron-exon junction or another SNP).
Linkage Mapping
A genetic map was drawn by connecting candidate SNPs after the filtering process to a hybrid of Capsicum frutescens cv. BG2814-6 x Capsicum annuum cv. NuMexRNaky RIL population (119 RILs) built at the University of California, Davis (https://pepchip.genomecenter.ucdavis.edu/).
Polymorphism Survey and Genetic Diversity
SNPs positioned in the linkage map were used to design locus-specific markers for the Fluidigm® EP1TM genotyping system. To validate their polymorphism and study the application of the SNP markers, 24 different C. annuum accessionss (CM334, YCM334, Tean, Yuwolcho, ECW, Bukang A line, Bukang C line, Lam32, Jeju, Dempsey, DRB, Perennial, 9093, ECW30R, Takanotsume, 35001, 35009, RS202, RS203, RS205, VK-515R, VK515S, LongSweet, AC2212) and three different C. chinense accessions (PI159236, Habanero, SNU11- 001) were employed. Genomic DNAs of each accessions were extracted by the hexadecyl trimethyl ammonium bromide (CTAB) method from young leaf tissues (Yang et al., 2012). Phylogenetic analysis was carried out using the neighbor-joining method in Darwin5 software (http:// darwin.cirad.fr/darwin, Perrier et al., 2003). Tree construction was based on the unweighted neighbor-joining method, and bootstraps were determined from 1000 replicates.
Results
Sequencing of Eight Hot Pepper Accessions and Quality Trimming
We produced a total of 4.1-4.4 Gb with 36-38 million reads from Jeju, LAM32 and Tean; a total of 7.1 Gb with 70 million reads from CM334; and a total of 3.5-3.6 Gb with 39-40 million reads from SNU-001, Yuwolcho, PI201234, and YCM334 (Table 1). After quality trimming with Sickle version 1.0, the collected data were reduced to 3.6-3.7 Gb with 35-37 million reads of 100-102 bp read length for Jeju, Tean, and LAM32; 5.3 Gb with 67 million reads averaging 79.5 bp for CM334; and 3.4-3.5 Gb with 39-40 million reads averaging 87-88 bp for SNU-001, Yuwolcho, PI201234, and YCM334 (Table 1).
Alignment of Each Accession to the Reference Sequence
The total acquired reference sequence of 21,665 kb was de novo assembled from 31,196 contigs derived from Bukang ESTs, with an average contig length of 696 bp. There was a total of 1,071 kb of interspersed repeat elements (5,216 elements). Unclassified repeats accounted for 626 kb of this. Of the remaining repeat sequence, the largest portion (278 kb) was annotated as long terminal repeats (LTR), which are composed of several hundred base pairs. Long interspersed nuclear elements (LINEs), which encode a reverse transcriptase (RT) and other proteins, accounted for 118 kb. In addition, there was 49 kb of DNA transposable elements, which tend to have short inverted repeats at each end. In addition to these interspersed repeat elements, 102 kb and 103 kb were marked as simple repeat and low complexity regions, respectively. Therefore, a total of 1,276 kb (5.9%) was masked as repeat sequences.
The trimmed reads of eight accessions (Table 1) were aligned to the reference sequence and the range of alignment ratio between each accession and the reference was 52.6- 70.5% (Table 2).
SNP Discovery and SNP Filtering
Sequences with read depth over 25x were used for SNP discovery, and those that were obviously distinguishable and different among accessions were defined as SNPs. Based on these criteria, a total of 58,151 SNPs were identified. From these, SNPs with only two types of genotypes and two alleles were filtered out. Among the remaining 57,502 SNPs, 33,315 were recognized as distinguishable in at least six different accessions (Table 3). In the next step, SNPs that showed a uniform segregation ratio were chosen. Among the 33,315 SNPs tested, 4,508 segregated 4:4 or 3:5 in the eight accessions. Finally, from those 4,508 SNPs, 1,910 were selected based on not having any other SNPs within 60 bp in either direction (Table 3).
Development and Validation of SNP Markers
Among 1,910 SNPs, a total of 1,282 were positioned on the hot pepper map produced by UC Davis. From these, we further selected 412 SNPs that showed clear and repeatable polymorphism, and that were evenly distributed over a ~3-cM interval in each chromosome (Supplementary Table 1). The linkage map showing the location of the 412 SNPs is presented in Fig. 1 and the SNP number per chromosome is given in Table 4. The entire length of the genetic map was 1,460.6 cM. There was an average of 34 SNPs in each linkage group, with the P1_Wild (177.1 cM) linkage group including the most SNPs. The P3 linkage group had the highest density (0.40 SNP/cM). The fewest SNPs mapped to linkage group P8_Wild. The number of SNPs per cM ranged from 0.16 to 0.40, with an average of 0.28 SNPs per cM. Accordingly, there an average of 1 SNP per 3.55-cM interval in the genetic map (Table 4 and Fig. 1).


To validate the 412 SNP markers, 24 C. annuum and 3 C. chinense accessions (Supplementary Table 2) were tested for diversity of pepper genotypes (Fig. 2). Between C. annuum accessions, the average number of SNPs was 143, ranging from 23 (ECW vs. ECW30R) to 205 (Takanotsume vs. YCM334). By contrast, between bell-type accessions of C. annuum (9003, Dempsey, ECW30R, ECW), the average number of SNPs was 51, ranging from 23 (ECW vs. ECW30R) to 69 (DRB vs. ECW). C. chinense accessions showed an average of 23 SNPs, ranging from 17 (Habanero vs. SNU11- 001) to 27 (Habanero vs. PI159236). The SNP number between C. annuum and C. chinense accessions averaged 179, ranging from 149 (VK-515S vs Habanero) to 209 (Dempsey vs PI159236). Overall, the average SNP number between all Capsicum accessions was 150, ranging from 17 (Habanero vs SNU11-001) to 209 (Dempsey vs PI159236). Based on the genetic similarity results, a cluster dendrogram placed the 27 pepper accessions into two main clusters and clearly differentiated each accession (Fig. 2). The first main cluster (I) comprised 15 different C. annuum accessions (9093, Dempsey, ECW30R, ECW, DRB, YCM334, RS205, RS203, RS202, Jeju, LongSweet, 35001, AC2212, CM334, Bukang A). The second cluster (II) consisted of 12 different accessions including seven C. annuum accessions (35009, Yuwolcho, Tean, Takanotsume, Bukang C, VK-515R, and VK515S), two wild species of C. annuum accessions (Perennial and Lam 32) and three C. chinenese accessions (PI159236, Habanero, and SNU11-001).
Discussion
Various approaches have been used to find SNPs in hot pepper, such as targeting sequences using COSII markers (Jung et al., 2010) or PCR using primers based on BAC sequences (Yang et al., 2009). Recently, ESTs have been widely used to find SNPs in crop varieties including tomato and barley because of their abundance and easy accessibility to the data (Kota et al., 2001; Labate and Baldo, 2005). The present work demonstrates that EST-derived SNP discovery using NGS is advantageous in hot pepper as well.
We prepared the reference sequence from Bukang ESTs using an approach similar to that described by Nicoli et al. (2012). However, we employed different SNP filtering methods to find valuable SNPs more effectively. By preparing a lengthy reference sequence after de novo assembly, we generated an alternative to WGS for reference sequence alignment to identify SNPs. Repeat areas are distributed throughout the genome in hot pepper. Therefore when preparing a reference sequence, repeat masking should be performed to find more accurate SNPs. In NGS for Jeju, LAM32, and Tean, parts of some reads contained low quality data and quality trimming resulted in small reductions in bases but not read number. However, for CM334, there was large amount of reduction due to low quality in both bases and reads. The sequences of SNU-001, Yuwolcho, PI201234, and YCM334 were high quality and did not show no much difference before and after trimming. Most accessions aligned to the reference with 50-70% of reads alignment. The variations might be due to the different genetic relation-ships between the accessions and the reference.
Overall, we identified many SNPs by aligning NGS sequence to the reference. However, the SNPs were not all equally valuable. Several factors can lead to false SNP findings, including base calling errors from NGS (Nielsen et al., 2011), miss-calls including overlapping genotypes (Anney et al., 2008), and false discovery of polymorphic SNPs that are actually monomorphic (Pettersson et al., 2008). To address this, we performed quality filtering of the SNPs. Our in-house SNP filtering process significantly decreased the SNP numbers: starting from 58,151 SNPs, the SNPs were filtered down to 1,910. After positioning the SNPs on the genetic linkage map, developing and testing SNP markers using the Fluidigm® EP1TM genotyping system, 412 SNP markers were finally selected and validated using 27 Capsicum accessions.
For MABC, background selection focuses on both reduction of donor segments around target genes and recurrent parent genome recovery. Successful MABC depends on genome size, population size, marker density in the map and the use of high throughput marker systems. Recently, Herzog and Frisch (2013) conducted computer simulations of MABC in genetic models of sugar beet, rye, sunflower and rapeseed, using the 10% quantile (Q10) value, the arithmetic mean and the standard deviation of the probability distribution of the proportion of recipient genome in the entire genome of selected individuals (as a percentage), for determining every backcross generation to measure recurrent parent genome recovery. Based on their data, the optimum designs, which minimize the required number of marker data points for target Q10 values of 96-99% in generation BC2 or BC3 in the model plants, employed marker densities of 2-20 cM intervals between markers and a population size of 50-100. In this study, the entire length of the genetic map covered by the SNP markers was 1,460.6 cM, suggesting that 73-730 markers (for 2-20 cM intervals) are needed for MABC in pepper. Our marker set produced an average of 150 SNPs between 27 accessions (corresponding to ~10 cM intervals), implying that our newly developed SNP markers should be useful for MABC between 27 accessions. SNP numbers between C. annuum and C. chinense were much higher than those between accessions from the same species such as bell types of C. annuum and C. chinense. This result implies that MABC could be more difficult in closely-related accessions than in distant accessionss. Despite this, our new SNP markers clearly distinguished 27 Capsicum accessions.
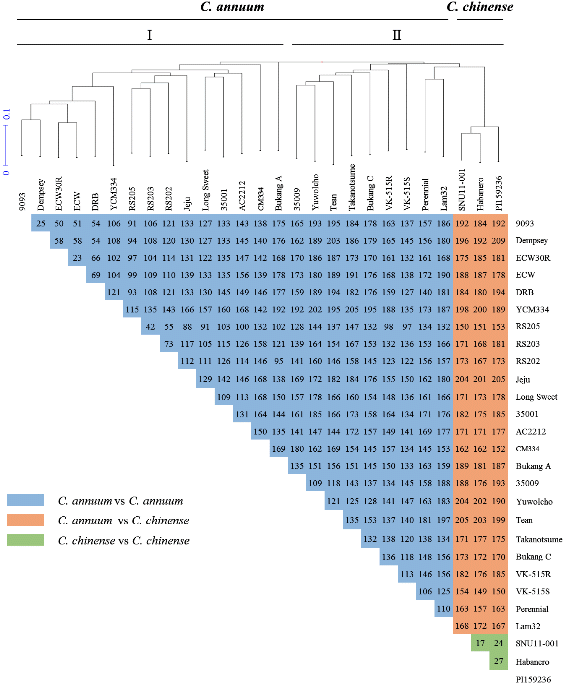
Among the accessions tested, AC2212 was positioned in the C. annuum cluster, rather than in the C. chinense cluster. AC2212 was originally classified as C. chinense based on agronomic information from the Centre for Genetic Resources, the Netherlands (http://applicaties. wageningenur.nl/applications/cgngenis/; Wahyuni et al., 2011). However, Wahyuni et al. (2013) reported out that AC2212 likely belongs to C. annuum instead of C. chinense, based on AFLP marker analysis. This result thus indicates that our newly developed SNP markers are accurate and can be used for the identification of diverse Capsicum accessions.
In conclusion, our SNP markers derived from transcriptome sequences will be valuable tools for MABC. In addition, the markers can be used for genetic mapping, comparative mapping and genetic diversity analyses in pepper and related species.
