

Introduction
Breaking the Genetic Code before Sequencing Technology: From Homopolymers to Ribosome-Bound tRNAs
First-Generation Sequencing Technology: From Maxam-Gilbert Sequencing to Sanger Sequencing
Second-Generation Sequencing Technology: Changing Paradigms by Pyrosequencing Technology Using Illumina, 454, Ion Torrent Sequencers
Third-Generation Sequencing Technology: Eager for Long-Read Sequences (PacBio and Oxford Nanopore Technology)
Discussion
Introduction
Research on DNA structure has been performed for more than 100 years, but relatively recently, we have come to know how DNA encodes genetic information. In 1868, Johann Friedrich Miescher found that there were a number of acidic phosphorus in the nucleus, and this nuclear material, which consists of protein and DNA, was called nuclein (currently, nucleic acid). In the late 1800s, Albrecht Kossel chemically analyzed nucleic acids and found four nitrogen bases: adenine, cytosine, guanine, and thymine (Pierce, 2012). In 1948, Erwin Chargaff and colleagues found that the ratio of DNA bases is consistent; the amount of adenine was the same as thymine, and the amount of guanine was always the same as cytosine (Chargaff, 2012). In 1952, Hershey and Chase revealed that genetic material in bacteriophages is DNA using 35S- and 32P-labeled protein and DNA, respectively (Hershey and Chase, 1952). In 1953, Watson and Crick discovered DNA double-helix structures (Watson and Crick, 1953). All the early studies have somehow contributed to the current sequencing technologies by providing the basis of DNA molecules. DNA sequencing accurately indicates and determines the order of nucleotides in a DNA molecule. Initially, an indirect method of determining the nucleotide sequence of RNA was used as a method for determining the sequence of bases in DNA. The method used homopolymers and ribosome-bound tRNA (Pierce, 2012). In 1977, the Maxam-Gilbert sequencing method was first developed based on the chemical modification of certain bases made by Allan Maxam and Walter Gilbert. Next, a chain terminal method that uses dideoxynucleotides was developed by Frederick Sanger. Sanger sequencing technology was predominant in the 1980s until next-generation sequencing (NGS) technology emerged in the mid-2000s. Furthermore, this new technology contributed greatly to the development of genome research by advancing techniques such as capillary electrophoresis, fluorescent labeling, and automated multiplexing. In 2001, the Human Genome Project was created using this technique. Since 2007, second-generation sequencing of various platforms from different companies has been developed to make genome research more approachable. Second-generation sequencing detects light signals that are released by pyrophosphate detaching during the DNA polymerization procedure. As a result, Roche’s 454, which uses the pyrosequencing method, or the various Illumina platforms, which use the sequencing by synthesis (SBS) method, have been developed. These technologies have made it possible to obtain high-throughput sequence data in a short time frame. However, the Illumina technology, which is by far the most popular platform in second-generation sequencing, generates short reads (100 to - 250 bp) and is not effective in conducting genome studies on new species with large or complex genomes (Schuster, 2007). Third-generation sequencing can produce long-read sequence data for a single molecule that can compensate for the drawbacks of using short-read sequencing data. Third-generation sequencing includes devices from Pacific Biosciences (PacBio) that use single-molecule real-time (SMRT) technology and those from Oxford Nanopore Technologies that use nanopore technology (Jain et al., 2018). Sequencing methods using these different platforms have been applied in various fields that use DNA sequences from species such as humans, plants, and animals. They can also contribute to the quality of human life in many different ways. In this article, the progress of sequencing technologies is divided into generations, and the principles and characteristics of each technology are discussed and summarized to determine which sequencing platform is suitable for different research purposes.
DNA sequencing accurately indicates and determines the order of nucleotides in a DNA molecule. Initially, an indirect method of determining the nucleotide sequence of RNA was used as a method for determining the sequence of bases in DNA. The method used homopolymers and ribosome-bound tRNA (Pierce, 2012). In 1977, the Maxam-Gilbert sequencing method was first developed based on the chemical modification of certain bases made by Allan Maxam and Walter Gilbert. Next, a chain terminal method that uses dideoxynucleotides was developed by Frederick Sanger. Sanger sequencing technology was predominant in the 1980s until next-generation sequencing (NGS) technology emerged in the mid-2000s. Furthermore, this new technology contributed greatly to the development of genome research by advancing techniques such as capillary electrophoresis, fluorescent labeling, and automated multiplexing. In 2001, the Human Genome Project was created using this technique. Since 2007, second-generation sequencing of various platforms from different companies has been developed to make genome research more approachable. Second-generation sequencing detects light signals that are released by pyrophosphate detaching during the DNA polymerization procedure. As a result, Roche’s 454, which uses the pyrosequencing method, or the various Illumina platforms, which use the sequencing by synthesis (SBS) method, have been developed. These technologies have made it possible to obtain high-throughput sequence data in a short time frame. However, the Illumina technology, which is by far the most popular platform in second-generation sequencing, generates short reads (100 to - 250 bp) and is not effective in conducting genome studies on new species with large or complex genomes (Schuster, 2007). Third-generation sequencing can produce long-read sequence data for a single molecule that can compensate for the drawbacks of using short-read sequencing data. Third-generation sequencing includes devices from Pacific Biosciences (PacBio) that use single-molecule real-time (SMRT) technology and those from Oxford Nanopore Technologies that use nanopore technology (Jain et al., 2018). Sequencing methods using these different platforms have been applied in various fields that use DNA sequences from species such as humans, plants, and animals. They can also contribute to the quality of human life in many different ways. In this article, the progress of sequencing technologies is divided into generations, and the principles and characteristics of each technology are discussed and summarized to determine which sequencing platform is suitable for different research purposes.
Breaking the Genetic Code before Sequencing Technology: From Homopolymers to Ribosome-Bound tRNAs
The easiest way to decode nucleotide sequences is to identify the nucleotide sequence of RNA and synthesize the protein by placing the RNA in a tube containing all the components necessary for protein biosynthesis (Pierce, 2012). This is an indirect method to sequence the RNA molecule by comparing amino acid sequences produced from the initial RNA molecules.
In 1961, the first method devised by Nirenberg and Matthai was to use synthetic RNA in the form of homopolymers made with polynucleotide phosphorylase (Martin et al., 1961). This enzyme does not need a template strand and randomly links RNA nucleotides. For example, when polynucleotide phosphorylase is added to a uracil nucleotide solution, it becomes an RNA molecule consisting of only polyuracil nucleotides. Next, polyuracil nucleotides are added to each amino acid tube labeled with a radioactive isotope. At this time, only radiolabeled proteins appeared in the tubes containing phenylalanine, indicating that the UUU codon designates phenylalanine. Similar results show that the CCC codon designates proline, and the AAA codon designates lysine (Matthaei et al., 1962).
In 1964, Nirenberg and Leder developed a technique using ribosome-bound tRNAs (Nirenberg and Leder, 1964). The short mRNA codons form a complementary base pair with the tRNA carrying encoded amino acids. The short mRNA deficient in ribosomes is mixed with tRNA and amino acids, and the blend is passed through a nitrocellulose filter paper. As a result, only the tRNA paired with the mRNA bound to the ribosome failed to pass through the filter paper. Next, more than 50 short mRNAs with known codons were synthesized and added to the ribosomes and tRNAs, respectively. After that, the mRNA and the tRNA bound to the ribosome were separated and it was determined which amino acid was present on the bound tRNA. Through these methods, the genetic code was completely decoded and provided the basis for genetic information analysis.
First-Generation Sequencing Technology: From Maxam-Gilbert Sequencing to Sanger Sequencing
First-generation sequencing can be divided into two methods based on different chain termination strategies. One is Maxam-Gilbert sequencing, which is based on different chemical treatments, and the other is Sanger sequencing, which is based on the enzymatic process with dideoxynucleotides (Fig. 1) (Hunkapiller et al., 1991).
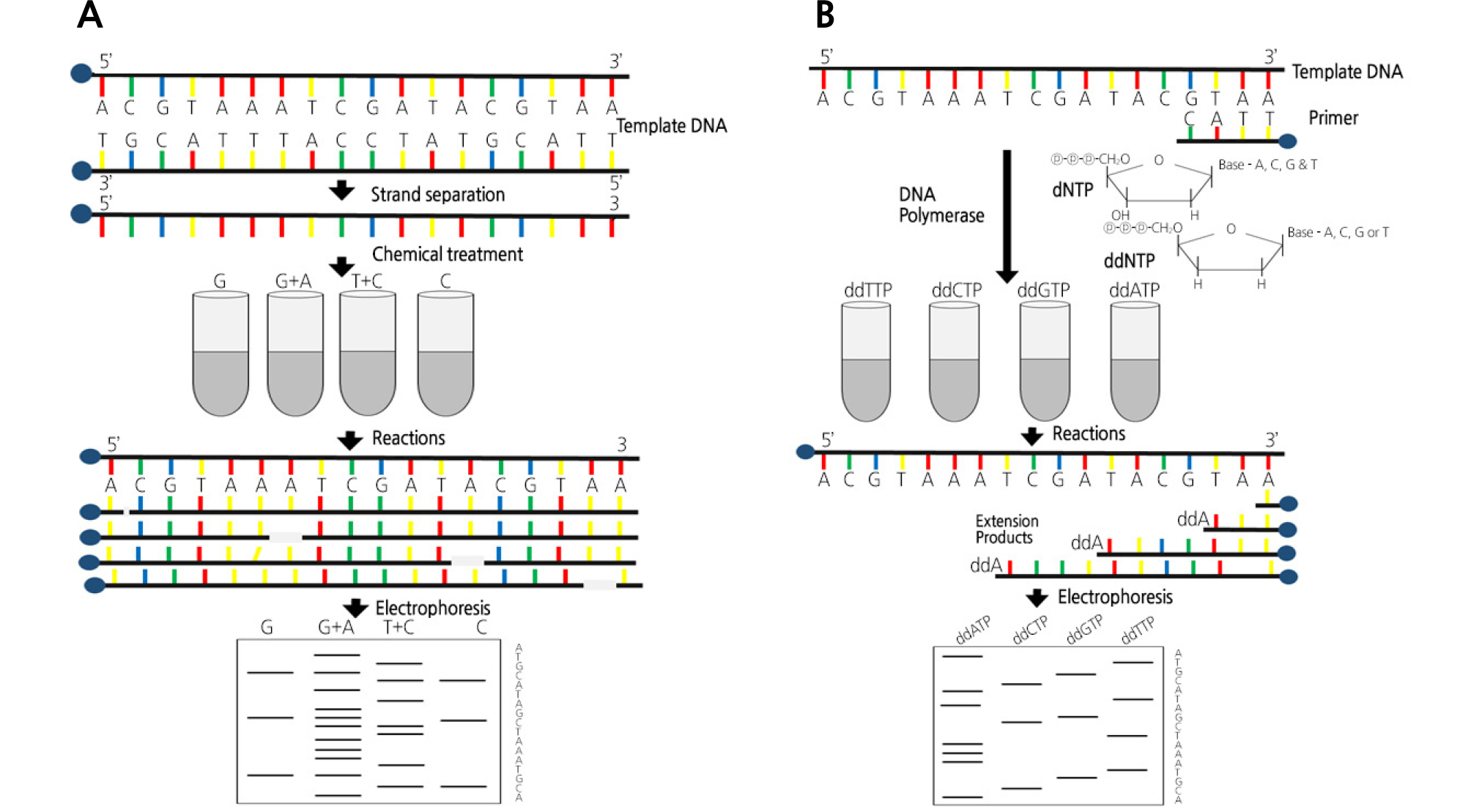
Maxam-Gilbert sequencing is a method based on chemical degradation. The basic principle is to randomly break down adenine, thymine, guanine, and cytosine positions using chemical agents specific to end-labeled DNA templates. Their products are identified using polyacrylamide gel electrophoresis (Maxam and Gilbert, 1977). First, DNA fragments are digested with restriction enzymes, which are then followed by the phosphorylation of the 5'-terminus using polynucleotide kinase and 32P-labeled ATP. The DNA fragment solution is divided into quarters and four kinds of chemicals are added. The chemical treatment takes place in four reactions, each of which cleaves at one or two nucleotide bases. For example, when formic acid is used in the first test tube, the purine base (A + G) is desulfurized. The use of dimethyl sulfate in the second test tube results in the methylation of guanine. In the third test tube, hydrazine is used to hydrolyze the pyrimidine base (T + C). The addition of sodium chloride to the hydrazine reaction in the fourth tube results in the only reaction of C in thymine. The addition of hot piperidine to the above reaction only cleaves the modified DNA. After the reaction is completed, four different solutions are subjected to gel electrophoresis and are then used for autoradiography to confirm the nucleotide sequences. However, currently, this sequencing method is rarely used, except for special purposes (França et al., 2002).
Sanger sequencing, developed by Frederick Sanger and also known as cycle sequencing technology, is a chain termination technique that uses dideoxynucleotides (dd-nucleotides). This technology is based on DNA polymerization, which is also used for polymerase chain reactions (PCRs). The single-stranded region of the DNA to be sequenced is used as a template, and a short oligonucleotide complementary to the template is used as a sequencing primer to initiate the synthesis (Sanger and Coulson, 1975; Sanger et al., 1977). When a partial amount of 2',3'-dideoxynucleoside triphosphates (ddNTPs) together with dNTPs (2'-deoxynucleoside triphosphates) is used in the DNA polymerization mixture, an extension of the strand is randomly terminated due to the absence of 3'-OH attached by ddNTPs. The dd-nucleotide has a hydroxyl group (-OH) at the 3' position of the ribose, which is a normal nucleotide group, substituted by the H group. During normal DNA synthesis, ddNTPs can also bind to the DNA chain. However, after entering the DNA chain, the ddNTPs no longer have an OH group at the 3' position of the sugar (the next nucleotide must bind), and the next nucleotide is no longer able to bind. As a result, the renal response is terminated. In the reaction, four different tubes are used. Each test tube contains dNTPs (dATP, dTTP, dGTP, and dCTP) that are components of DNA templates in common. Each test tube contains a different ddNTP chain terminator. For example, a G reaction tube contains ddGTP/dNTPs, and each chain produced in a test tube is terminated when the ddGTP binds randomly. Through this process, chain polymerization can be completed at all G points in the G test tube, and a series of mutually different DNAs are produced for each test tube (Metzker, 2005).
However, in the early Sanger method, the DNA fragments generated by the reaction must be separated by electrophoresis in a polyacrylamide slab gel and read out radioactively. Therefore, the complicated manipulation process requires a lot of time, money, and labor. The development of devices such as the ABI Prism 3700, in which the Sanger method is combined with capillary electrophoresis and fluorescence labeling agents (BigDyeTM), can be performed in a shorter time than before. This is because all four reacting tubes can be combined into one due to the different labeling colors of the four bases. The development of these machines has helped many types of research, such as the Human Genome Project, until the NGS method was introduced (Hutchison III, 2007).
The first genomic sequencing of plants was performed using the Sanger method on rice and Arabidopsis (Li and Harkess, 2018).
Second-Generation Sequencing Technology: Changing Paradigms by Pyrosequencing Technology Using Illumina, 454, Ion Torrent Sequencers
Unlike the Sanger method, second-generation sequencing produces a lot of sequence data. It is also called deep sequencing technology because it can generate multiple copies of the same genomic locations. The second-generation sequencer amplifies the DNA sequence, though there is a slight difference depending on the model, of which Roche’s 454 and Life Technology’s Ion Torrent use emulsion PCR, while Illumina’s models use solid-phase amplification (Metzker, 2010).
Roche sequenced the human genome with 454 technology for the first time in 2008. This result, which was completed in 4.5 months using NGS, received considerable attention compared to the Human Genome Project, which took 13 years. The 454 sequencer enables a read length of 400 bp and produces 35 Mb of data with a run time of 10 hours. It fixes one DNA strand in one bead and amplifies it with emulsion PCR (emPCR) (Dressman et al., 2003). At the end of the amplification, each bead is covered with the same DNA sequence, which is replicated a million times. Each bead also enters a well in the device called the PicoTiterPlate and starts sequencing reactions. The 454 sequencing technique is also called pyrosequencing chemistry because it uses the light signal from the pyrophosphate released by the chain elongation. In turn, the released pyrophosphate is used to generate adenosine triphosphate (ATP) and adenosine phosphosulfate (APS). The generated ATP and luciferate generate light by converting luciferin to oxyluciferin (Rothberg and Leamon, 2008). DNA fragments amplified by DNA polymerase are attached to a microbead and are sequenced by decoding the fluorescence signal generated by the subsequent DNA amplification process. The long read, one of the major features of the 454 series, is known to favor de novo sequencing, which assembles reads without reference sequences, but has the disadvantage that the amount of data produced in one run is relatively small compared to other NGS platforms (Ronaghi, 2001).
Second, Illumina introduced a technology called sequencing by synthesis (SBS). In this method, an adapter oligonucleotide is ligated to a shortly fragmented genomic DNA and then the oligonucleotide is hybridized to a complementary primer with an adapter sequence immobilized on the surface of a glass flow cell. In this state, when the PCR reagent is added, the DNA immobilized on the free primer that exists on the peripheral surface is bent, and the other adapter is combined and amplified (a bridge amplification process) (Fig. 2). With the proper dilution, a single DNA fragment is spatially well separated, and a cluster that originated from the initial DNA fragment is formed (Mardis, 2013). This cluster is a clone that has the same aggregate originating from a single DNA fragment and acts like a microbead in emulsion amplification. Once the clusters are formed on the flow cell, the sequencing primer specific for the adapter sequence initiates the sequencing reaction using SBS technology (Lim et al., 2011). Illumina has developed various NGS instruments based on the SBS method, and it is still the most predominant technology among the second-generation platforms. Illumina’s NGS series produces a large amount of data with low run prices overall and is therefore widely used in large-volume sequencing research (Table 1). The Illumina platform has a much lower error rate and an overall accuracy of over 99.5%. This technology can be applied in various ways, such as whole-genome sequencing (WGS), exome sequencing, ChIP-seq, and genotype-by-sequencing (GBS) (Head et al., 2014; Chung et al., 2017). However, there is a tendency for substitution errors and limitations for decoding AT- and GC-rich regions (Caporaso et al., 2012).
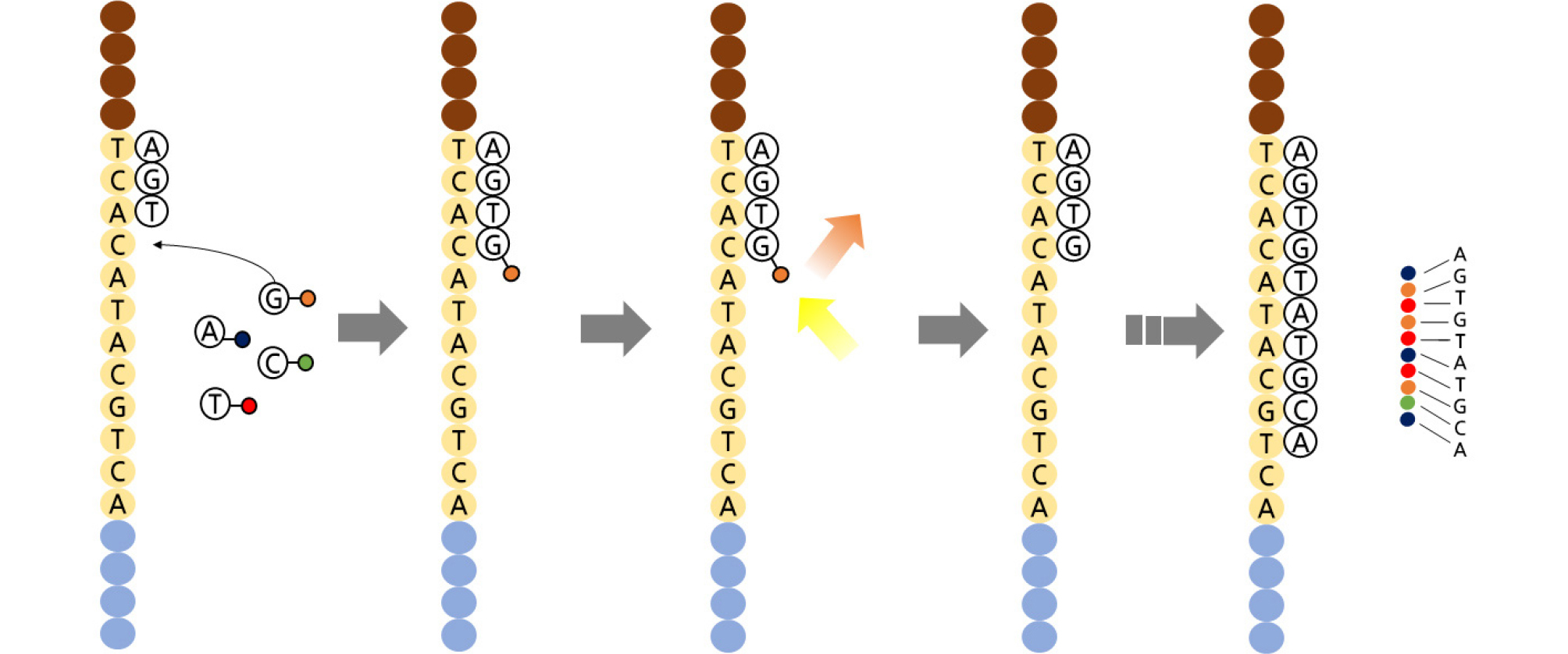
Table 1. Comparison of various second-generation sequencing platforms
Finally, Life Technology’s Ion Torrent also adopted emPCR, like the 454 platform, followed by SBS sequencing. Ion Torrent’s sequencing is generally performed in semiconductor chips. Different template DNAs enter separate wells, and an ion sensor is attached to the bottom of each well to read the signal changes (Merriman et al., 2012). Like other NGS instruments, it first amplifies the template on the bead in an emPCR mixture and prepares the template on each bead surface. According to the cyclic sequencing method, A, T, G, and C nucleotides cause synthesis reactions. When a nucleotide is polymerized into DNA, the hydrogen ions released as a by-product are captured by an electrical signal and the base is read (Van Dijk et al., 2014). When the same nucleotide repeats, a large signal is caught and distinguished. Since the fluorescence is not used, the entire camera shooting process is eliminated, and the equipment becomes simple and compact. Consequently, fluorescent markers were no longer needed, and reagent costs were greatly reduced (Metzker, 2010).
Third-Generation Sequencing Technology: Eager for Long-Read Sequences (PacBio and Oxford Nanopore Technology)
Third-generation sequencing has been developed to overcome the use of short-read sequences, which is a disadvantage of the second-generation sequencing techniques. This generation of sequencing is different from the second-generation technology in that the PCR amplification process is omitted and DNA single molecules (mostly high molecular weight DNA) can be sequenced as they are. Third-generation sequencing techniques include SMRT technology developed by PacBio and Oxford nanopore technology (ONT) based on nanopore methods (Table 2) (Jain et al., 2018).
Table 2. Comparison of various third-generation sequencing platforms
PacBio developed single DNA molecule sequencing called SMRT technology (Fig. 3) (Eid et al., 2009). SMRT technology uses high molecular weight (HMW) DNA as a template and synthesizes it with polymerase reactions. The nucleotide sequence is determined in real time by detecting the reaction occurring at every base by a fluorescence pulse change. There is only one molecule of DNA polymerase attached at the bottom of the sequencing chip (Rhoads and Au, 2015). Here, a sequencing reaction with the template DNA fragment is performed, the reaction is detected in real time, and the base sequence is read. A fluorescent molecule attaches to the end of the nucleotide’s phosphate, and when the base-binding reaction occurs, the molecule is dropped, and the fluorescence pulse stops. The machine detects these signal changes and uses a CCD camera like second-generation sequencers. The sequence data obtained through this method are 90 Mb in length, with the advantage that the read length is very long (20 kb on average) (Quail et al., 2012).
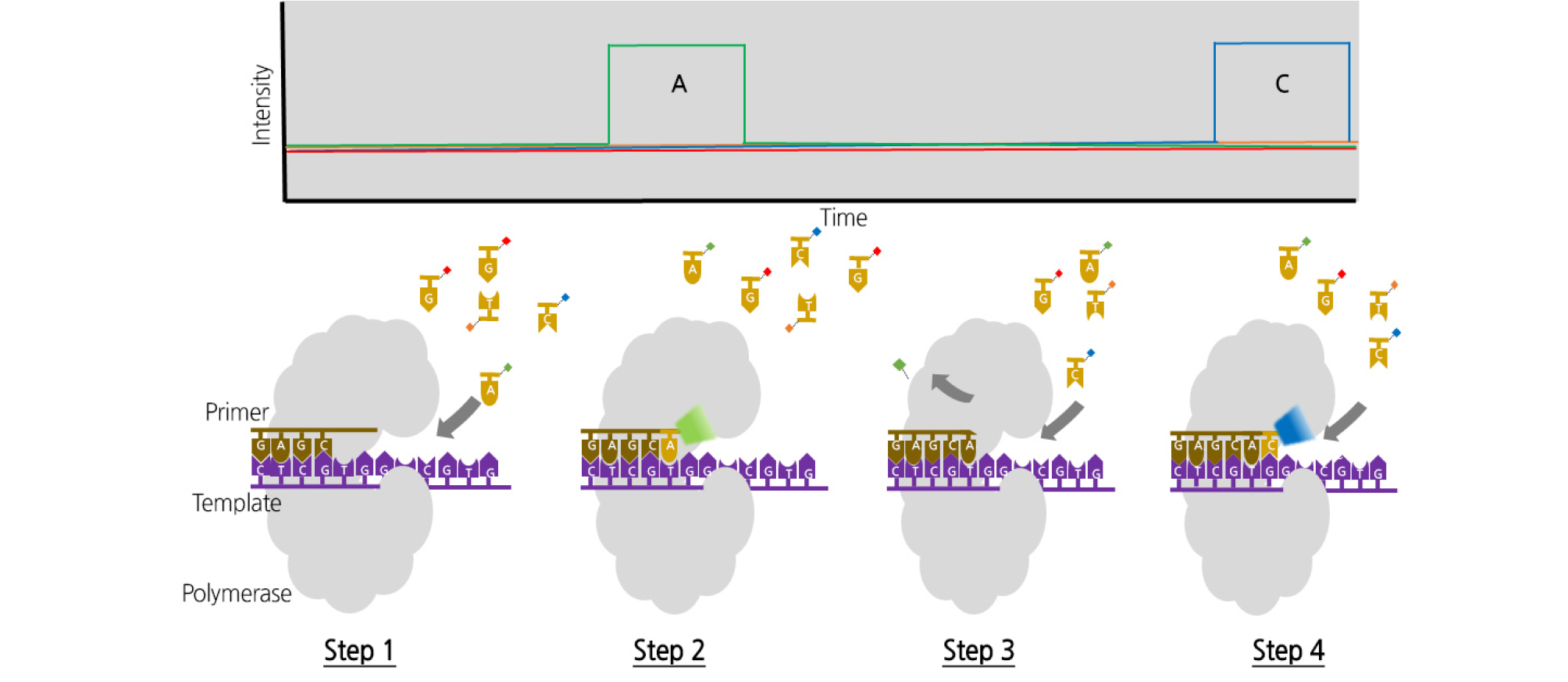
Fig. 3.
SMRT DNA sequencing. (a) The DNA polymerase immobilized on the bottom of the zero-mode waveguide (ZMW) binds to a single molecule of the DNA template. (b) (1) At the active site of the polymerase, the phosphor-linked nucleotides form a cognate association with the template. (2) The fluorescent output increases in the corresponding color channel. (3) The formation of a phosphodiester bond extricates the dye-linker-pyrophosphate product and diffuses from the ZMW, stopping the fluorescence pulse. (4) The polymerase moves to the next position and the next cognate nucleotide binds the active site to initiate the next pulse.
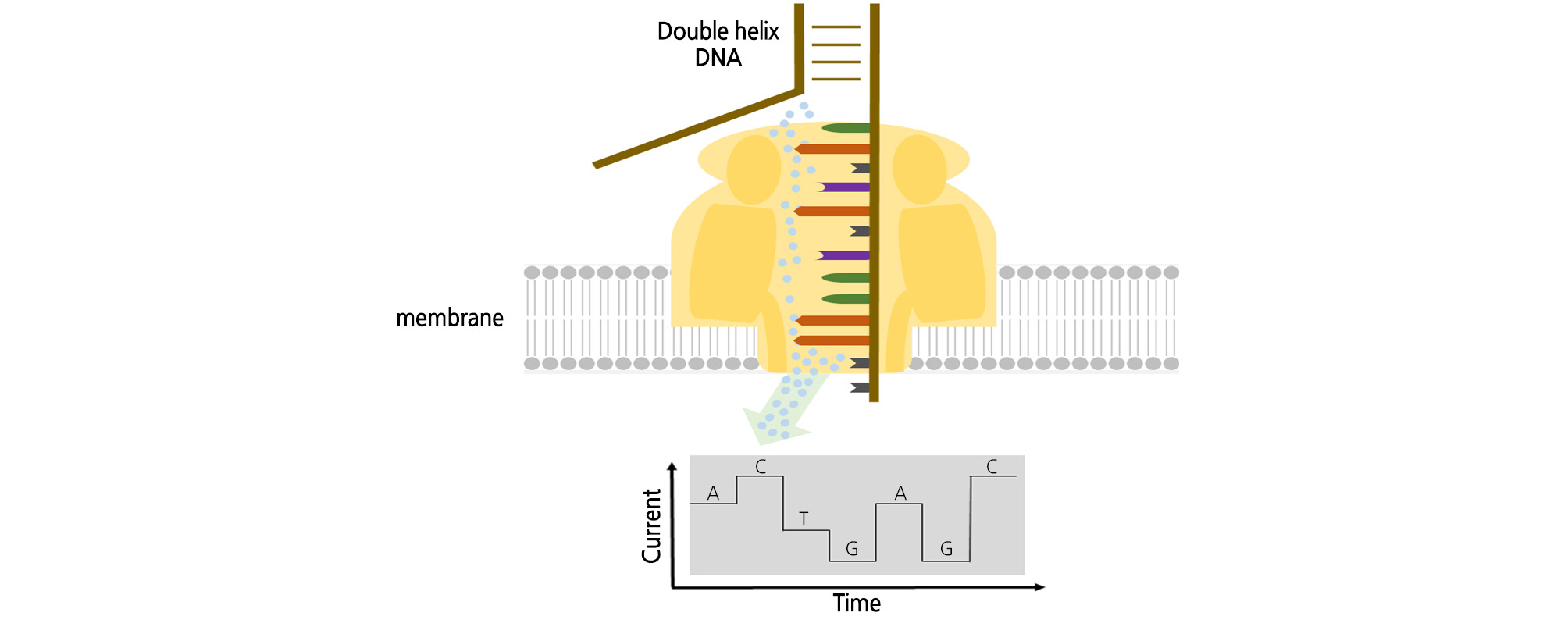
ONT is a single DNA molecular sequencing method that discards the fluorescence method of NGS or PacBio and reads the base signal by electric current (Fig. 4) (Reuter et al., 2015). This nanopore sensing technology uses an artificial tube called nanopore, which is a current path. When a nucleotide passes through a nanopore, it interrupts the current flow. The A, T, G, and C nucleotides have different current flows (Branton et al., 2008). The nucleotide sequence is determined using this potential change. Nanopores are proteins made up of membranes, and proteins that form pores are common. α-Hemolysin serves as an ion and molecular channel in the cell membrane and as a protein pore of heptamer with an inner diameter of 1 nm and the same size as a single molecule of DNA. Pores are made on a synthetic film made of SiNx or SiO2. Ion or electron beams can be precisely matched to the molecular structure. It can be hybridized with proteins to increase the specificity; graphene is a good candidate material (Bleidorn, 2016). The base sequencing method is an exonuclease sequencing method that reads out the types of nucleotides that are liberated by cleaving the nucleotide of the template instead of receiving the signal of the DNA synthesis in the template. The exonuclease is bound to the α-hemolysin molecule with nanopore and attached to the cyclodextrin. When exonuclease cleaves the nucleotide in the template, it passes through the pore and specifically interrupts the current flow to read each nucleotide sequence (Leggett and Clark, 2017). ONT is an innovative ultra-small sequencer that eliminates both the PCR amplification and fluorescence imaging processes. Because of the array chip principle, high-throughput systems are possible.
Discussion
Sequencing technology has evolved over many generations in a variety of ways. Researchers have been running the Human Genome Project for more than 10 years with Sanger sequencing, but it now performs human genome analysis in just a few days. Second-generation sequencing allows us to obtain a large amount of sequence information in a short period and to analyze it using various bioinformatics tools. However, due to the short-read sequence limit of second- generation sequencing, it has become difficult to analyze the genomes of new species. The development of third-generation sequencing, which produces a long-read sequence, has overcome the limitations, but it has the disadvantage of being more expensive than second-generation sequencing. Using the features of various platforms of multiple generations of sequencing enables the technology to be applied to different fields of biological research. For example, in agricultural science, breeders spend a lot of time genotyping thousands of individual plants included in the different populations by using many molecular markers such as random amplified polymorphic DNAs, amplified fragment length polymorphisms, restriction fragment length polymorphisms, and simple sequence repeats. All the conventional molecular markers demand complicated and time-consuming experimental procedures such as PCR, gel electrophoresis, and even handling dangerous radioactive materials. In particular, most of the procedures require previous knowledge of genomic sequence data to obtain appropriate genotyping information. Newly developed genotyping methods, such as GBS, exome sequencing, and RAD-seq, have made it possible to use single-nucleotide polymorphisms, which are theoretically infinite in a genome, thanks to the deep sequencing technologies offered by the Illumina sequencing platform. The newly developed methods have significantly reduced time, cost, and labor for genotyping and have enabled breeders to construct ultra-density genetic/quantitative trait locus maps. The technical progress not only has contributed to genotyping accuracy but has also increased the overall efficiency of plant molecular breeding by applying a number of molecular markers linked to different agriculturally important traits. In addition, a variety of in silico tools are being developed to combine data generated by multiple sequencing methods. Genetic information can be easily obtained by developing the sequencing, and it can be applied to various fields based on this information. In the future, we will be able to obtain better quality sequence data, which will contribute to the development of genomics by applying them to relevant downstream analyses.
